





علائم را بشناسید، بیماری را تشخیص دهید
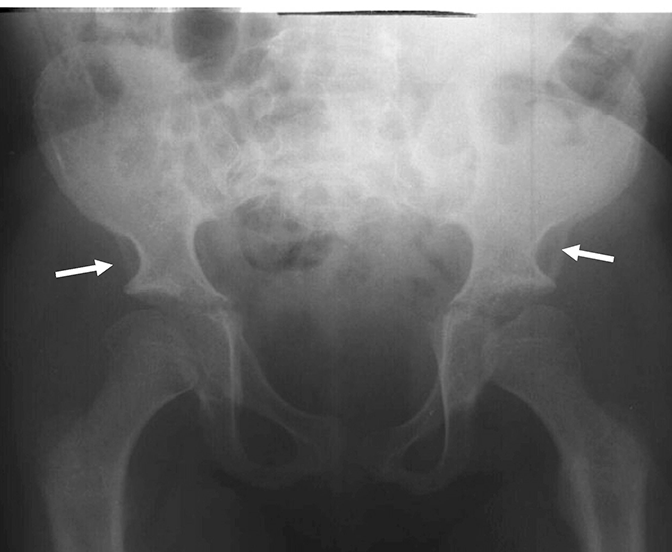
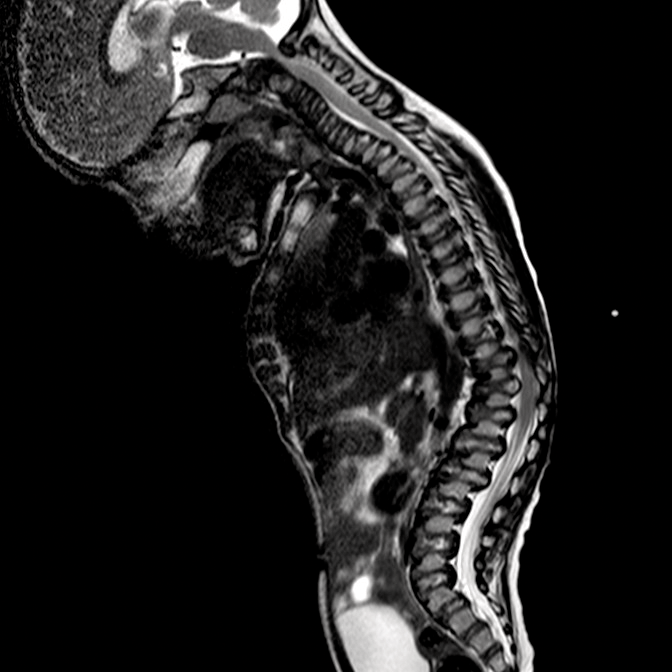
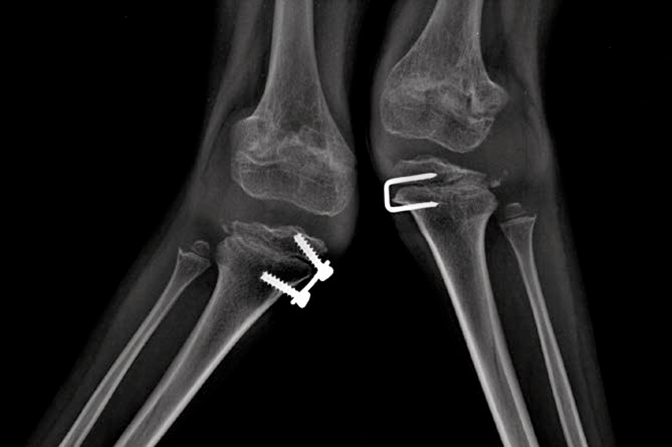
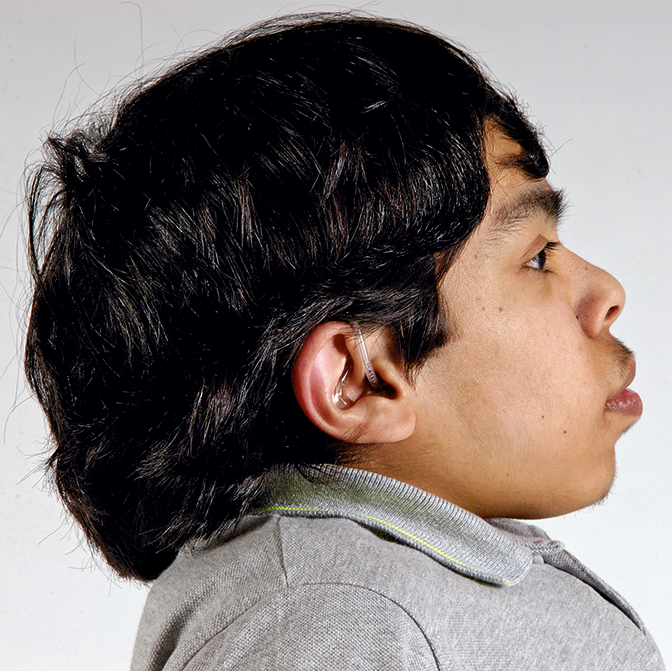
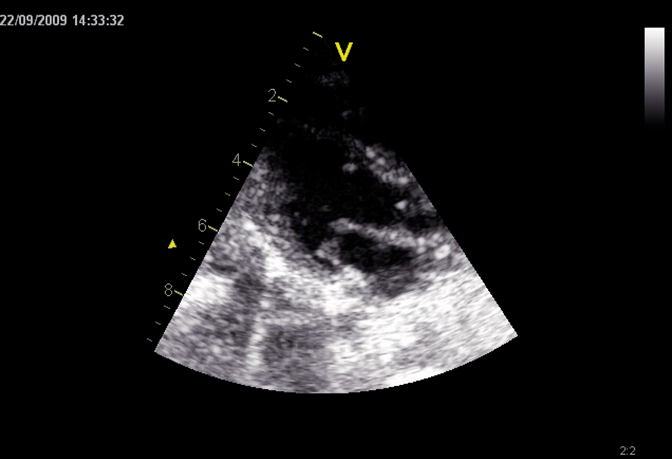
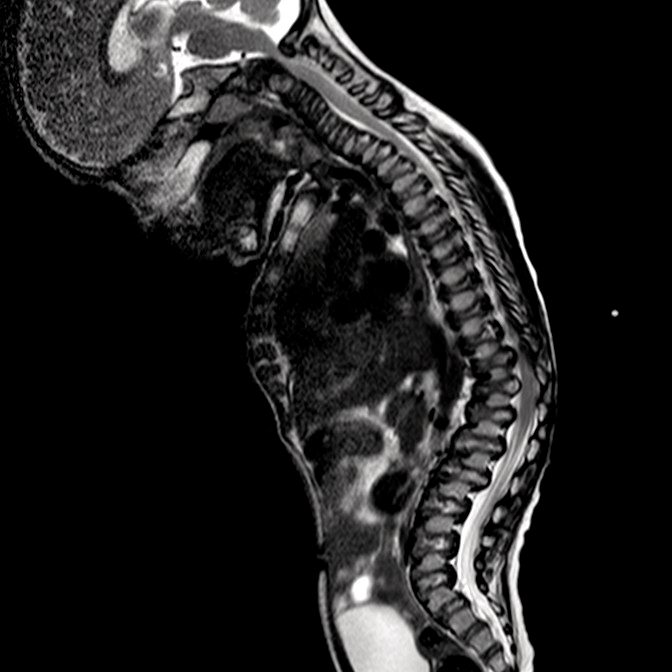
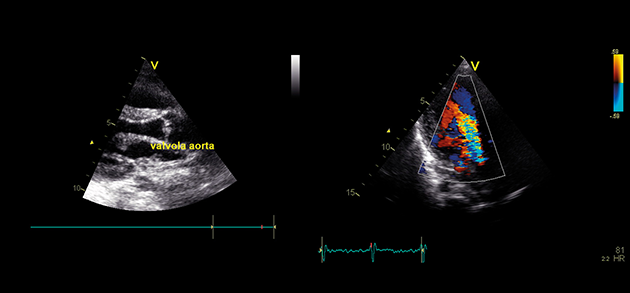
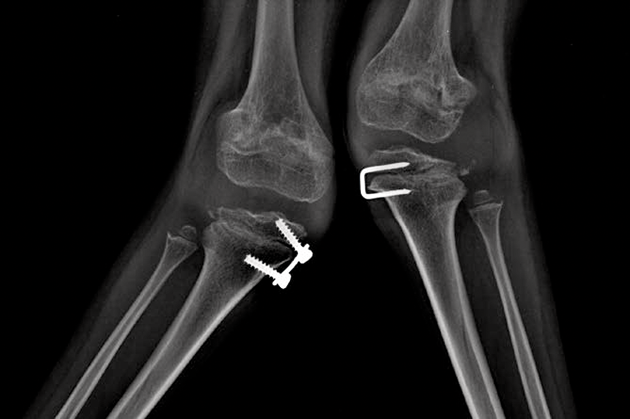
اختلالات موکوپلی ساکاریدوز (MPS) گروهی از اختلالات جدی پیش رونده، ارثی و ژنتیک می باشند که بر سیستم ها و کارکردهای مختلف بدن اثر می گذارند و حاصل کمبودهای ناهمگن آنزیمی هستند.1-3 تنوع در تظاهرات بیماری و روند پیشرفت آن – و نیز برداشت کلی جامعه پزشکی از نادر بودن آن – باعث شده که MPS در بخش های تخصصی کمتر تشخیص داده شود و منجر به تأخیر در تشخیص و بالا رفتن احتمال بروز عواقب مخرب آن گردیده است.4،5
بهینه سازی عوارض بلندمدت بیماران و کمک به شروع زودهنگام درمان با شناختن علائم MPS و ارجاع دادن فوری بیماران مشکوک به متخصصان ژنتیک یا مراکز بیماری های متابولیک. ارجاع به مراکز بیماری های متابولیک نخستین گام مهم در تشخیص افتراقی و شروع زودهنگام درمان است.4
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}