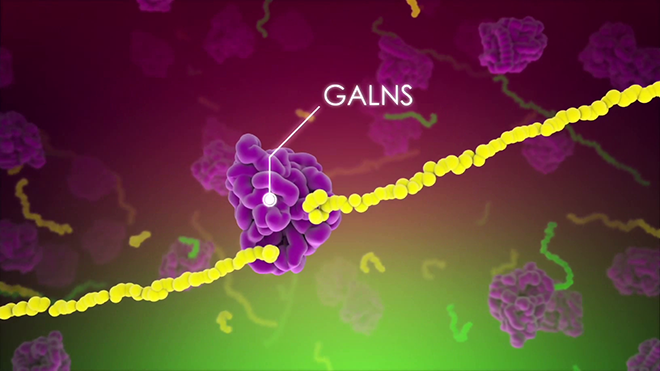
MPS: مخفی شده در دید کامل
شناسائی زودهنگام در گروی شک زودهنگام می باشد
اختلالات MPS از لحاظ بالینی ناهمگن هستند و فارغ از سرعت پیشرفت که از کند تا سریع متغیر است، منجر به عوارض جدی می شوند. اگرچه نرخ پیشرفت تنها جزو خصوصیات کلی شدت بیماری هستند اما بیماران دارای سرعت پیشرفت کند بیماری هم در معرض خطر مریضی های جدی و مرگ و میر معمول مرتبط با بیماری پیش رونده سریع هستند.2,6,14
- بسیاری از افراد مبتلا به بیماری وخیم در صورت عدم درمان به سن بزرگسالی نخواهند رسید (دهه دوم و سوم)6،15
- علائم در افرادی که نوع پیش رونده کند بیماری را دارند معمولاً دیرتر در طول زندگی شروع خواهد شد14,16
بیماران مبتلا به MPS ممکن است الگوی کلاسیک یا غیرکلاسیک علائم و نشانه ها را داشته باشند.16
- نشانه شناسی کلاسیک بصورت الگوهای براحتی قابل تشخیص علائم و نشانه هاست که بطور مشروح در متون علمی و در روال های بالینی به آنها پرداخته شده است16
- نشانه شناسی غیرکلاسیک ظرفت بیشتری دارد – بدون ویژگی های بارز چهره، کوتاهی قامت یا درگیری های آشکار عضلانی استخوانی انواع کلاسیک MPS – در نتیجه تمایز دادن این فنوتیپ با سایر اختلالات رایج تر استخوانی یا متابولیک دشوار است16
- بعنوان مثال در یک مطالعه اخیر عنوان شده است که حدود 25% بیماران مبتلا به سندروم مورکیو نوع MPS IVA) A) دارای فنوتیپ غیرکلاسیک هستند17

بیماران غیرکلاسیک

بیماران غیرکلاسیک
بیماری پیش رونده کند و/یا ظاهر فنوتیپیک غیرکلاسیک می تواند با سیر بالینی وخیم در پیشرفت های اندام های خاص اشتباه گرفته شود.18 فارغ از فنوتیپ، علائم می توانند تا آسیب نهائی اندام پیش بروند.12
علائم آشکار و بطور کلی قابل مشاهده باید شک بیماری را بوجود بیاورند
فارغ از محیط بالینی، ممکن است علائم آشکار و بطور کلی قابل مشاهده ای وجود داشته باشند که باید شک بیماری را بوجود بیاورند. با معاینه بیشتر امکان دارد بواسطه ارزیابی های بالینی تخصصی، یافته های آزمایشگاهی و تاریخچه بیمار، نشانه های بیشتری کشف کرد. این تقسیم بندی در شکل زیر نشان داده شده است.
علائم و نشانه های MPS1،2،5،11،12،16-30
خصوصیات عمومی
- راه رفتن غیرعادی
- دیسپلازی استخوانی
- محدودیت باز شدن انگشتان
- خصوصیات زمختی چهره
- درد مفاصل
- ماکروسفالی
- پکتوس کاریناتوم
- کاهش استقامت بدنی/عدم توانائی در ورزش
- قامت کوتاه/کندی رشدa
خصوصیات آشکار شونده با ارزیابی های تخصصی
- راه رفتن غیرعادی
- بدشکلی های استخوانی
- دیسوستوزیس مالتی پلکس
- پای ضربدری
- درگیری مفاصل (انقباض، شل بودن مفاصل) بدون التهاب
- سابلاكسشن ستون فقرات
خصوصیات عمومی
- کاهش تحرک مفاصل
- خشکی و درد مفصل ران
- درد مفاصل
- خشکی یا شل بودن مفاصل
خصوصیات آشکار شونده با ارزیابی های تخصصی
- سندروم کارپال تونل
- درگیری مفاصل بدون التهاب مفاصل یا آسیب های فرساینده استخوانی
خصوصیات عمومی
- ضعف شنوائی هدایتی و/یا حسی عصبی
- بزرگ شدن زبان
- عفونت گوش میانی
خصوصیات آشکار شونده با ارزیابی های تخصصی
- دریچه نای غیرعادی
- پل بینی فشرده شده
- بزرگ شدن غده لوزه حلقی
- بزرگ شدن لوزه
- مواد مخاطی در گوش میانی
- تنگ شدن مجاری هوای سوپرا گلوتیک و اینفرا گلوتیک
- بدشکلی استخوانچه های شنوائی
- ترشحات مکرر و بیش از حد بینی
- عفونت مکرر گوش میانی
- فشردگی/ضخیم شدن نای
- انسداد لوله ای
- ضخیم شدن غشای پرده صماخ
خصوصیات عمومی
- آب مروارید
- کدورت انتشاری قرنیه
- گلوکوم
خصوصیات آشکار شونده با ارزیابی های تخصصی
- تاربینی
- کدورت قرنیه با ظاهر بارز "شیشه مات"
- دوربینی شدید
- هیپرترولیسم
- ناهنجاری های عصب بینائی (التهاب و آتروفی)
- پیشروی پیرامونی عروق به داخل قرنیه
- خروج کاذب و پیش رونده تخم چشم
- کاهش تیزی بینائی
- رتینوپاتی
- دوبینی
خصوصیات عمومی
- ناهنجاری های رفتاری (معمولاً در MPS IVA و VI مشاهده نمی شود)
- تأخیر رشد (معمولآً در MPS IVA و VI مشاهده نمی شود)
- ضعف شنوائی
- حملات صرع (معمولآً در MPS IVA و VI مشاهده نمی شود)
خصوصیات آشکار شونده با ارزیابی های تخصصی
- کیست های عنکبوتی (معمولاً در MPS IVA و VI مشاهده نمی شود)
- آتروفی مغز (معمولاً در MPS IVA و VI مشاهده نمی شود)
- سندروم کارپال تونل
- فشردگی/میلوپاتی/سابلاكسشن طناب نخاعی
- بزرگ شدن فضای پره واسکولار
- هیدروسفالوس
- دیسپلازی دندانی
- پاکی مننژیت گردنی
- ادم پاپی/آتروفی نوری
- ناشنوائی حسی عصبی
- ناهنجاری های شدت-سیگنال
- تنگی کانال نخاعی
- ونتریکولومگالی
خصوصیات عمومی
- کاهش استقامت بدنی/عدم توانائی در ورزش
خصوصیات آشکار شونده با ارزیابی های تخصصی
- فشار خون ریوی
- ضخیم شدن، نارسائی، یا تنگ شدن دریچه های میترال یا آئورت همراه با بزرگ شدن عضلات بطن چپ
- نارسائی دريچه سه لختى
خصوصیات عمومی
- کاهش استقامت بدنی/عدم توانائی در ورزش
- آپنه خواب
خصوصیات آشکار شونده با ارزیابی های تخصصی
- انسداد مجاری هوای فوقانی و تحتانی (تنگ شدن نایژه ها، تنگ شدن مجاری هوای سوپرا گلوتیک و اینفرا گلوتیک)
- کاهش پیش رونده حجم ریه
- عفونت های تنفسی
- اختلالات خواب (آپنه خواب انسدادی/سندروم وقفه تنفسی و سندروم مقاومت مجاری هوای فوقانی)
خصوصیات عمومی
- دل درد
- یبوست
- بزرگی کبد و طحال
- فتق
- شل بودن مدفوع
خصوصیات آشکار شونده با ارزیابی های تخصصی
خصوصیات عمومی
- ناهنجاری سطوح گونه ای
- نقص در ساخت دندان ها
- هيپودنشيا
- تیز شدن دندان های نیش
- بیلچه ای شکل شدن دندان های ثنایا
- نازکی مینا
خصوصیات آشکار شونده با ارزیابی های تخصصی
- ناهنجاری سطوح گونه ای
- نازکی مینا
aدرگیری استخوانی و کوتاهی قامت شاید در برخی بیماران کمتر مشهود باشد.
از آنجائیکه تظاهرات استخوانی در میان افراد مبتلا به MPS رایج است، این اختلالات MPS شاید شبیه به دیگر عارضه های استخوانی باشند.1،10،16 MPS باید در محیط های بالینی مختلف، اعم از موارد زیر در نظر گرفته شود:16،30
- کلینیک های دیسپلازی استخوانی
- ارتوپدی
- روماتولوژی اطفال
- کلینیک های متابولیک استخوانی
ناتوانی های شناختی و ذهنی جزو علائم اصلی و ممیز تمام اختلالات MPS نیستند. هرگز نباید به دلیل عدم وجود ناتوانی های شناختی/ذهنی احتمال MPS را رد کنید.1
ارجاع و تشخیص به موقع بهترین شانس بهبود زندگی بیماران است.1،6 علائم اصلی بالینی MPS را بشناسید و بیماران مشکوک را فوراً برای آزمایشات تشخیصی به متخصصان ژنتیک یا مراکز بیماری های متابولیک ارجاع دهید.1،16،30