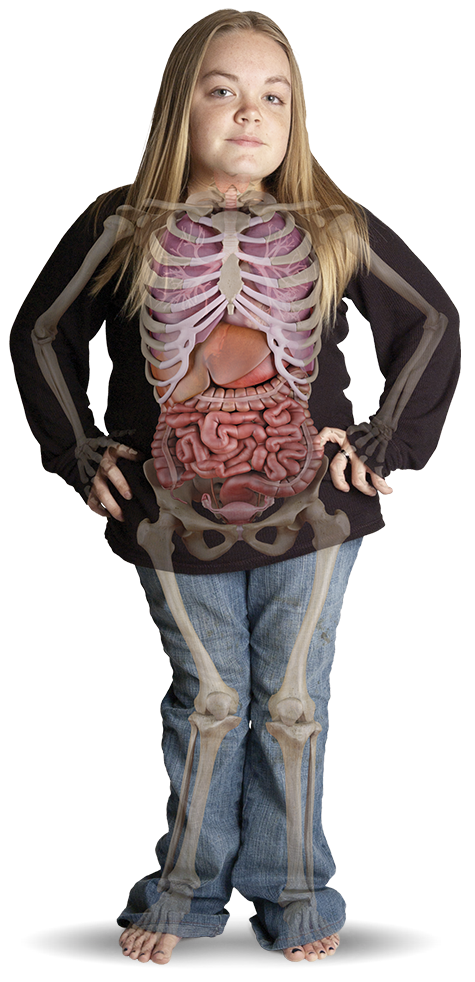
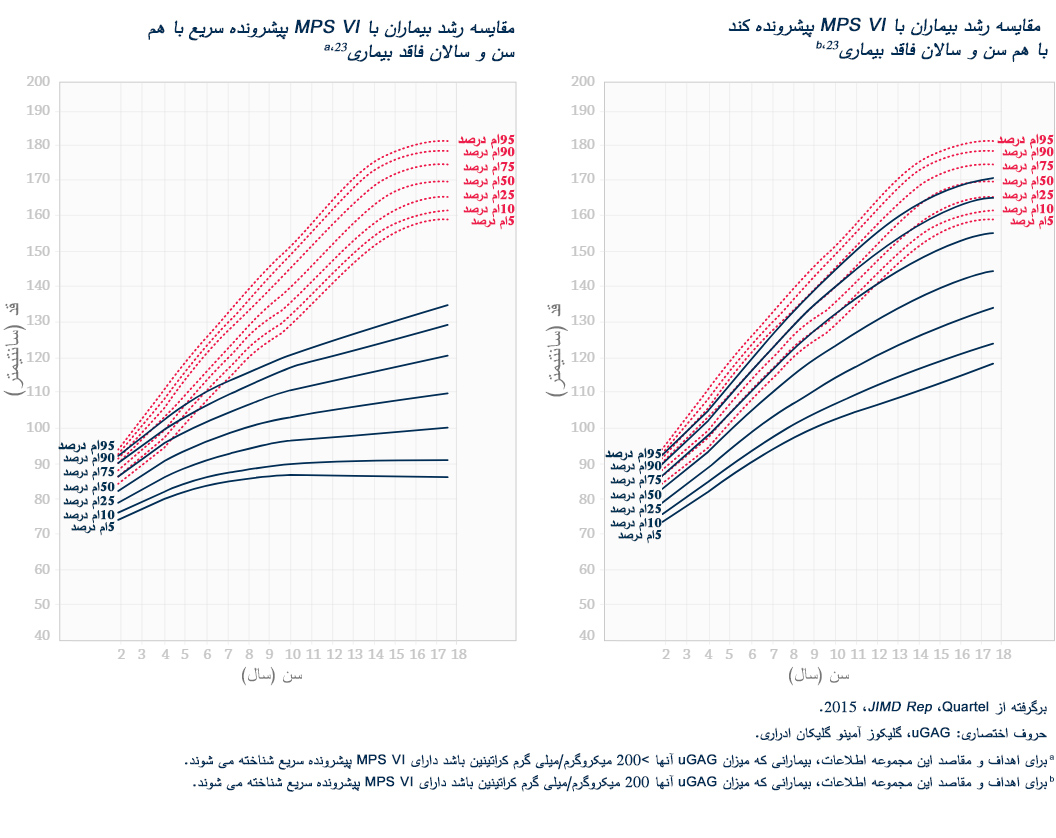
تشخیص دقیق اختلالات MPS هیچگاه تا این حد اهمیت نداشته است
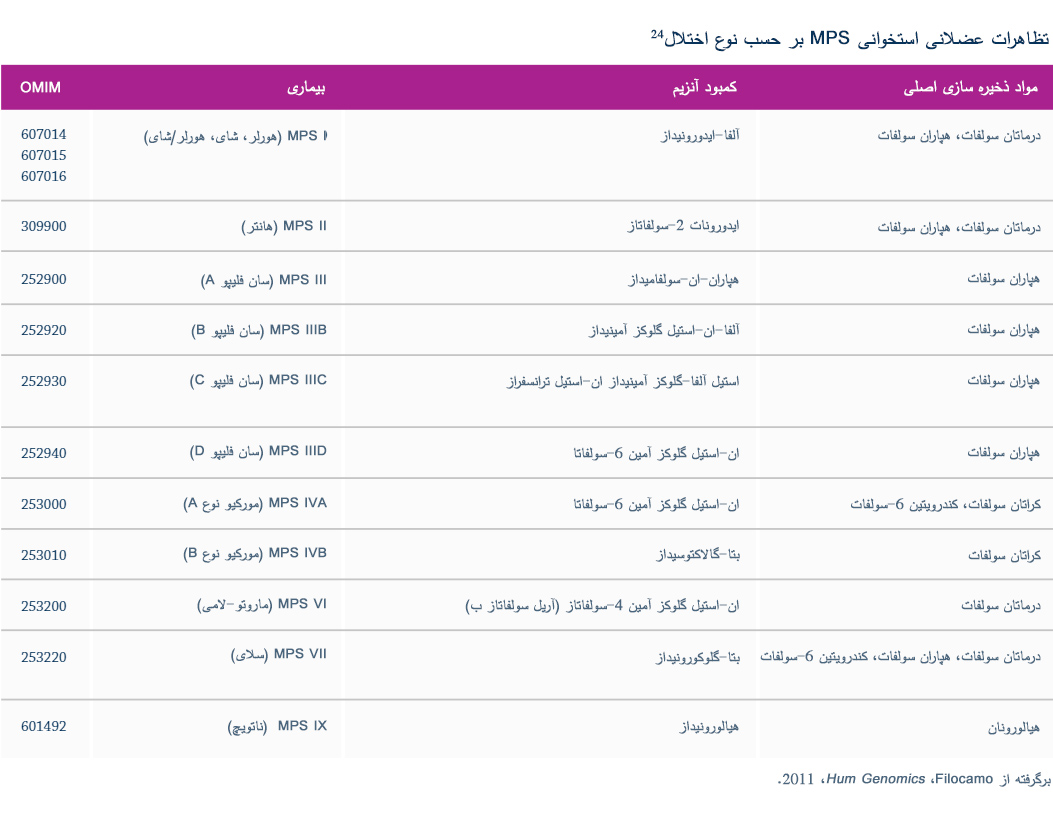
شناسائی زودهنگام کمبودهای آنزیمی خاص و دست یافتن به تشخیص قطعی می تواند در صورت وجود درمان جایگزینی آنزیم (ERT) زمینه را برای مداخله زودهنگام فراهم آورد.2،24،25 حتی در صورت عدم وجود امکانات ERT، مدیریت های خاص بیماری می تواند عواقب و عوارض را برای بیماران بهینه سازد.2 در صورت شک به MPS باید یک آزمایش جامع آنزیمی انجام شود.1 شناسائی کمبودهای آنزیمی منوط به سنجش عملکرد آنزیمی در فیبروبلاست ها یا لوکوسيت هاست که شیوه توصیه شده تأیید تشخیص می باشد.26 شرح کلی مسیر تشخیصی MPS VI در ادامه آورده شده است.
نمونه آزمایش تشخیصی برای MPS VI
شک بالینی و/یا رادیوگرافی به MPS VI
صحبت درباره آزمایش با خانواده
انتخاب یک گزینه غربالگری
- در غربالگری می توان از آزمایش DBS استفاده کرد؛ استاندارد طلائی فعلی برای تشخیص، نمونه های فیبروبلاست یا لوکوسيت می باشد
- آزمایشات مرکب برای غربالگری چندین اختلال لیزوزومی در حال حاضر وجود دارند
- برای رد احتمال کمبود آنزیم های مختلف و مشکلات انتقال لیزوزومی نیاز به سنجش آنزیم های متعدد است
- برای تأیید صحت نمونه باید آنزیم لیزوزومی مرجع نیز در همان زمان سنجیده شود
- زمان حمل نمونه خون باید به حداقل ممکن کاهش داده شود؛ تحویل در روز بعد توصیه می شود
- اگر سطح عملکرد ASB پایین باشد، باید برای رد احتمال چندین کمبود سولفاتاز یک بار دیگر سولفاتاز سنجیده شود
- می تواند شک را ایجاد کند اما تشخیص MPS را رد نمی کند
- uGAGها در بیماران دارای بیماری پیش رونده کند می توانند طبیعی یا فقط کمی بالا باشند
- امکان دارد بالا بودن uGAGها در نمونه های ادراری رقیق شده تشخیص داده نشود
- باید کیفی و کمی باشد: ارزیابی کمی برای تعیین مقدار کل uGAG و ارزیابی کیفی برای درماتان سولفات
- نخستین تخلیه صبحگاهی 10 تا 20 میلی لیتر ایده آل است
- فرستادن ادرار بصورت منجمد یا روی کاغذ فیلتر خشک شده
- به واحدها و محدوده های مرجع توجه داشته باشید
- مقایسه نتایج آزمایشات uGAG از آزمایشگاه های مختلف شاید کار صحیحی نباشد چون امکان دارد از شیوه های متفاوتی برای آزمایش استفاده نمایند

آزمایش تشخیصی برای MPS VI
سنجش عملکرد ASB در نمونه های فیبروبلاست یا لوکوسيت (استاندارد طلائی)
- پایین بودن سطح عملکرد ASB
- برای تأیید صحت نمونه باید آنزیم لیزوزومی مرجع نیز در همان زمان سنجیده شود
- اگر سطح عملکرد ASB پایین باشد، باید برای رد احتمال چندین کمبود سولفاتاز یک بار دیگر سولفاتاز سنجیده شود
- در صورت استفاده از نمونه فیبروبلاست، باید برای رد احتمال بیماری سلول-I آنزیم مرجع مانوز 6 – فسفات اندازه گیری شود
گزینه های تکمیلی آزمایشات
- تأیید کم بودن سطح عملکرد آنزیم ASB با تکرار اندازه گیری روی نمونه یا بافت جداگانه
- در صورت امکان پذیر بودن، شناسائی 2 موتاسیون پاتوژنیک در ژن آریل سولفاتازB (ARSB) (1 از هر والد)
یک الگوریتم پایه نشان دهنده آزمایشات که می تواند برای تأیید تشخیص مورکیو نوع (A (MPS IVA استفاده شود در زیر آورده شده است.
الگوریتم تشخیص مورکیو نوع A27 
آزمایشات آنزیمی برای بررسی پایین بودن سطح عملکرد تمام 11 آنزیم وجود دارند. این شیوه می تواند خیلی سریع و دقیق سطح عملکرد آنزیم های کلیدی را برای چندین اختلال MPS و اختلالات ذخیره سازی لیزوزومی مورد ارزیابی قرار دهد.1،26