آیا یک بیماری نادر ژنتیک در بخش شما پنهان شده است؟
MPS که اغلب با اختلالات دوران کودکی یا بیماری های عضلانی استخوانی اشتباه گرفته می شود شاید آنقدر که تصور می کنید نادر نباشد
اختلالات موکوپلی ساکاریدوز (MPS) گروهی از کمبودهای آنزیمی با تظاهرات ناهمگن و پیشرفت متغیر بیماری هستند که احتمال داده می شود 1 مورد از هر 22500 مورد کودک تازه متولد شده در جهان را تحت تأثیر قرار دهد.1،2
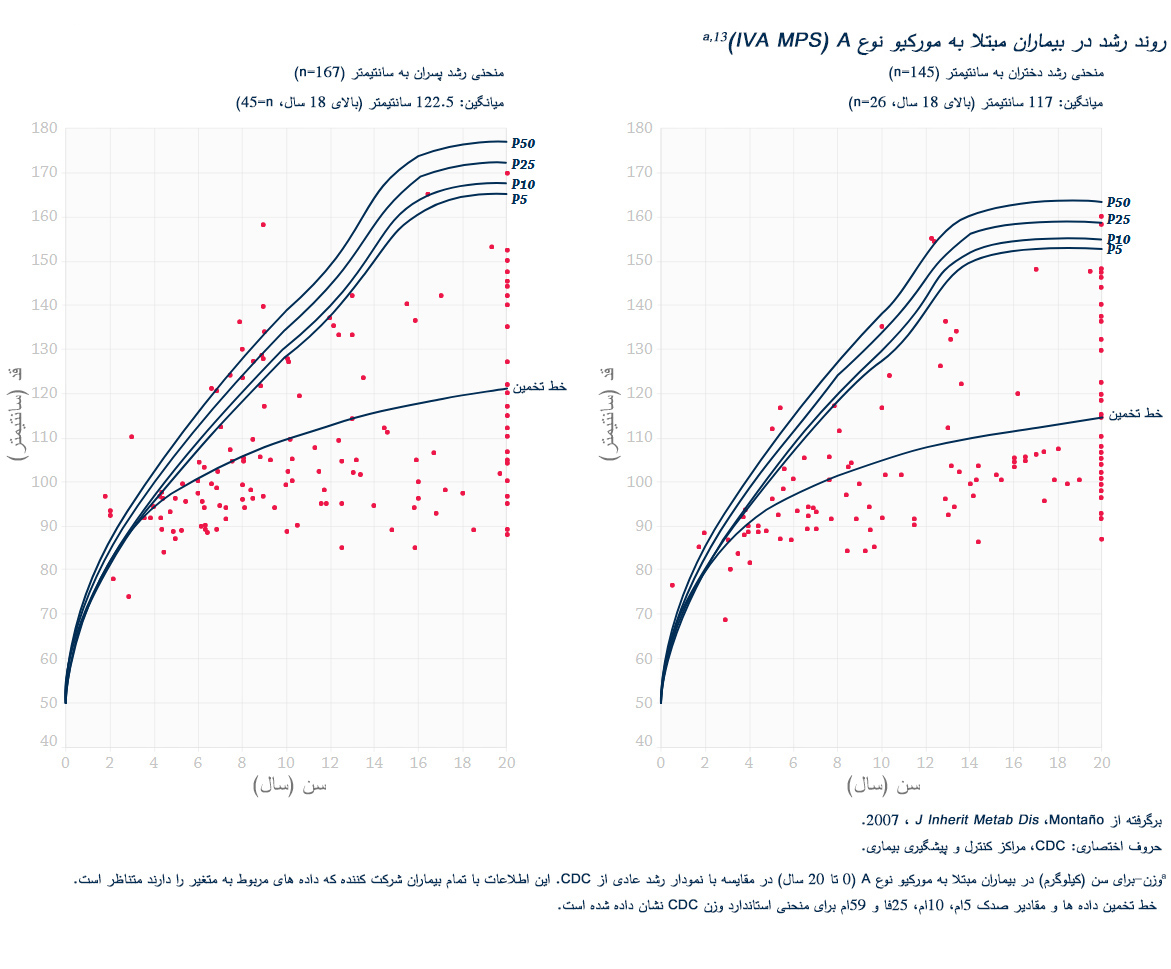
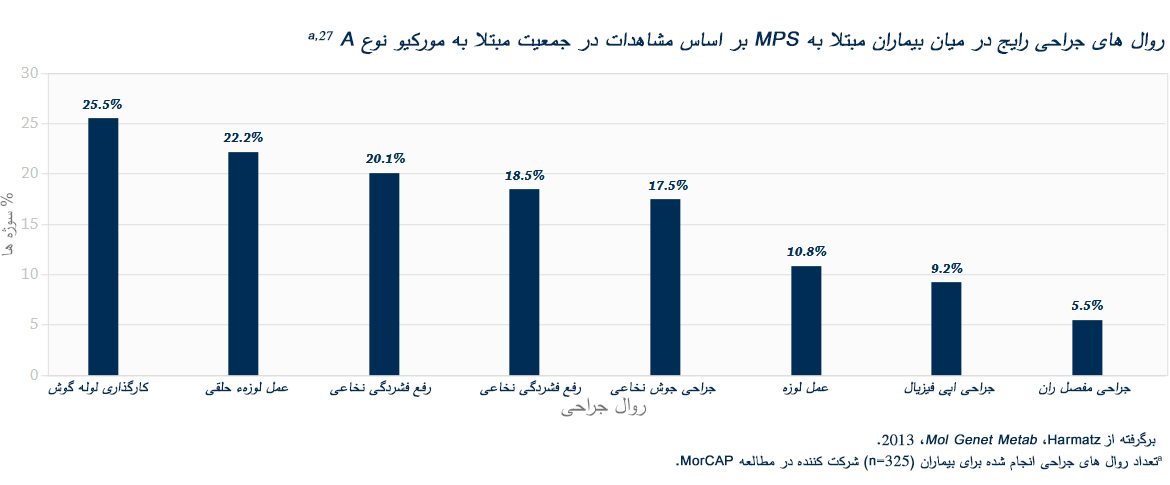
علائم بالینی و سرعت های پیشرفت مشابهی در میان تمام اختلالات MPS دیده می شود. الگوهای پیشرفت بیماری MPS که در بیماران مبتلا به MPS VI دیده شده است در تصاویر زیر به نمایش گذاشته شده است.
پیشرفت متغیر بیماری در بیماران مبتلا به MPS VI
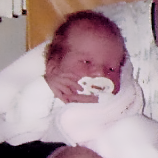



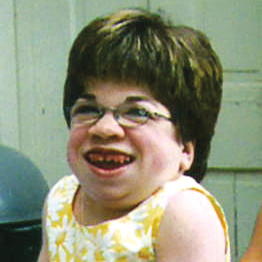
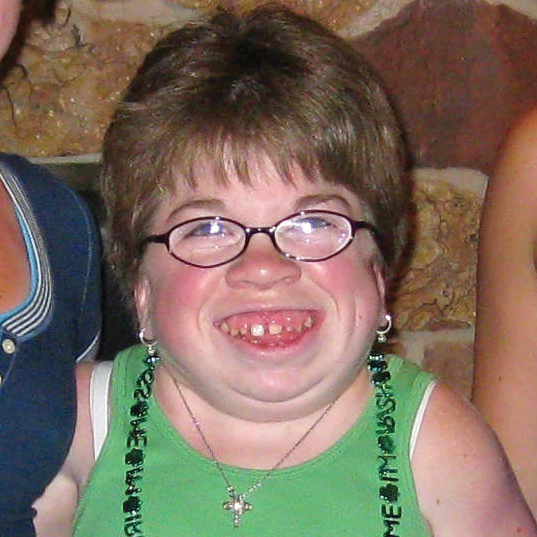
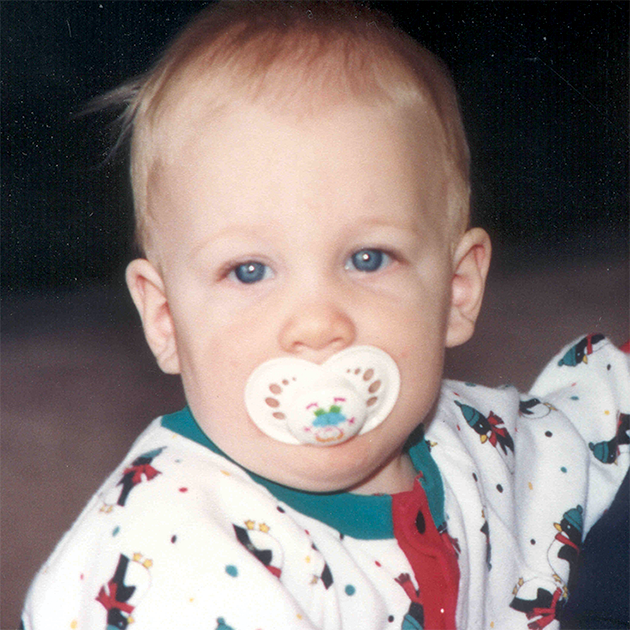




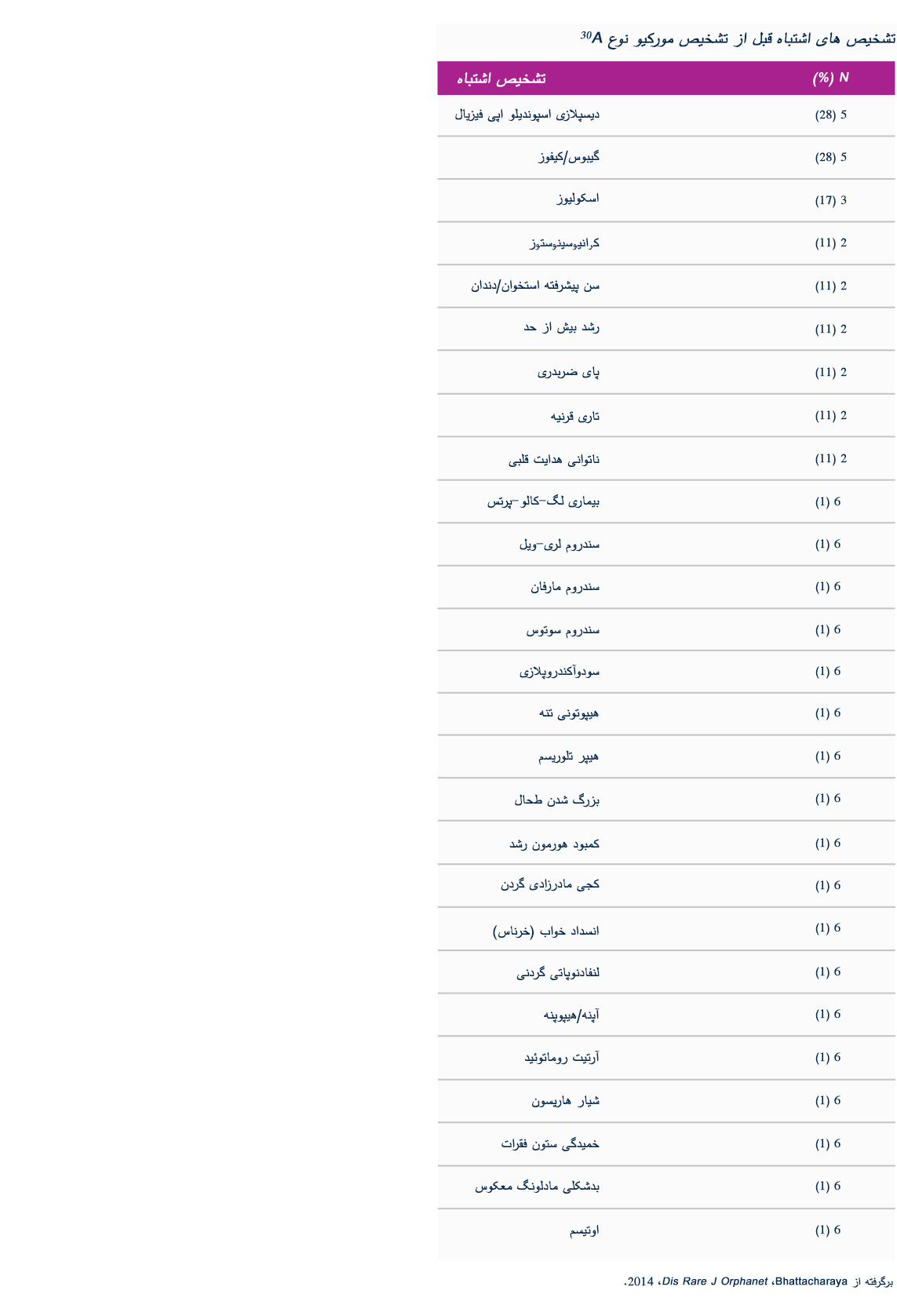
تأخیر در تشخیص بسیار شایع است و می تواند تبعات مخربی برای بیماران داشته باشد3
تصور نادر بودن بیماری، تنوع در روند پیشرفت بیماری و تظاهرات آن و نیز مجموعه علائم غیرمشخص مرتبط با MPS تشخیص را دشوار می سازند. زمان رسیدن به تشخیص از بروز نخستین علائم می تواند از شش ماه تا چند دهه متغیر باشد.3

تشخیص زودهنگام برای بهینه سازی عوارض بیماران از اهمیت بالائی برخوردار می باشد3،5
تشخیص زودهنگام می تواند بواسطه فراهم کردن دسترسی به امکانات مدیریت خاص بیماران و درمان جایگزینی آنزیم (ERT) عوارض را برای بیماران بهینه سازی نماید.3،5–8
برای اکثر اختلالات MPS یک ERT وجود دارد یا در حال توسعه می باشد. بهترین راه برای تشخیص MPS و شروع درمان ارجاع دادن بیمار مشکوک به یک متخصص ژنتیک یا مراکز بیماری های متابولیک آشنا با آزمایشات مخصوص آن می باشد.9
مراقب علائم و نشانه ها اولیه MPS، بخصوص این موارد باشید:3,10–12
- کلاسیک
- فتق
- ناهنجاری های مفاصل و استخوان ها
- کوتاهی قامت
- هرگونه تغییر تدریجی و پیش رونده در ظواهر جسمانی
- غیر کلاسیک
- تظاهرات ظریف ناهنجاری های استخوانی، شامل ضخیم شدن دنده ها یا ترقوه، شامل پای ضربدری ظریف، و ناهنجاری یا درد مفاصل
- کاهش ظرفیت ورزش و/یا درگیری بدون توجیه قلبی عروقی، بخصوص سوفل قلب در اطفال
- ضعف های بدون توجیه شنوائی یا بینائی (کدورت قرنیه)