


Supportive clinical evidence from sibling studies suggests that early intervention provides multiple opportunities to improve patient outcomes through disease-specific management and early initiation of ERT, if available.1-6
ERT, whether initiated early or later in life, has been shown to improve key clinical parameters, such as endurance and pulmonary measures, which are critical to quality of life, maintenance of ambulation, and activities of daily living.7,8
You are now leaving the MPSReference website. Please choose a link below to be redirected to more information on specific available enzyme replacement therapies.
Vimizim.com for treatment of Morquio A
Naglazyme.com for treatment of MPS VI
For additional information on clinical trials, visit www.clinicaltrials.gov.
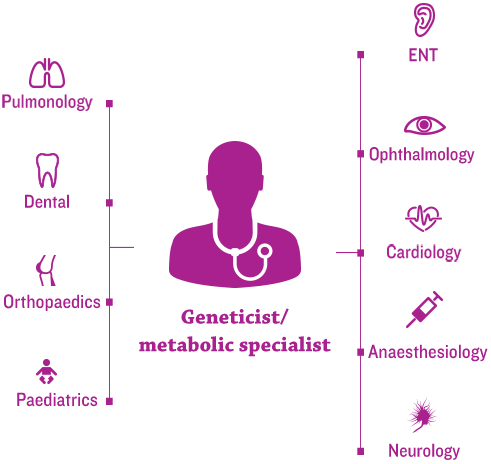
The new era of management for progressive, complex, genetic conditions, such as mucopolysaccharidosis (MPS) disorders, hinges on the efficient coordination of each patient’s healthcare team by a medical home.1
Geneticists and/or metabolic specialists are typically at the centre and help to coordinate multidisciplinary care and an individualised management plan.2,3
Cardiologists play a critical role in multidisciplinary coordinated care of patients with MPS for several reasons:2,4
Because of the prevalence and severity of cardiac involvement, continued cardiac assessment is a key component to effective management of these patients.3,4 The most common cardiac manifestations of MPS that should be monitored include the following:5
Many MPS disorders have available management guidelines and
speciality-specific consensus recommendations regarding lifelong management of MPS. Guidelines typically recommend the following:3,6
Early and ongoing assessments from a coordinated care team can improve patient outcomes and may help prevent irreversible damage.6
While cardiac involvement is seen in all MPS subtypes, most studies show it tends to occur earlier and more frequently in MPS I, II, and VI than in MPS III and IV. However, it is important to remember that signs and symptoms are unpredictable and clinically heterogeneous both across and within MPS subtypes. Patients may exhibit non-classical and/or classical cardiac signs of MPS, as well as rapidly or slowly progressing disease.4 This underscores the importance of both baseline and ongoing cardiac monitoring in all patients with MPS.3
A number of expert reviews are available that present state-of-the-art practices in the assessment and monitoring of cardiac abnormalities in patients with MPS.3,7-9 Many MPS disorders also have available management guidelines and cardiology-specific consensus recommendations regarding chronic care.3,6,10
These reviews and guidelines include the following overarching recommendations:3,6,10
The figure below details recommended diagnostic techniques and assessments to evaluate cardiac anatomy and function in patients with MPS at both initial diagnosis and regular intervals thereafter. Recommended assessment intervals range from 1 to 3 years, depending on MPS subtype.4

Additional routine cardiac evaluations should be considered before major operative interventions, including careful auscultation of heart and lungs, as well as right arm and leg blood pressure measurements.4
Cardiologists involved in the ongoing management of patients with MPS should keep the following in mind:4
Frequency of assessments and involvement of specific specialists vary across the different MPS types. For patients with MPS diseases associated with primary neurodegenerative and cognitive complications, such as MPS I, II, and III, additional and regular neurobehavioural and psychiatric evaluations are recommended.6,11,12
In addition to speciality-specific assessments that should be done to facilitate positive long-term outcomes for patients with MPS, important steps can be taken by the coordinating physician, typically the geneticist and/or metabolic specialist, related to general health. Their role in educating other healthcare professionals (e.g. dentists, physiotherapists, paediatricians, family doctors) and families about the disease and general management strategies is critical and should include the following:
Speciality-specific assessments, as well as regular physical examinations and overall health interventions, should follow recommended guidelines, which may vary among MPS subtypes.3
Improvements in the treatment of MPS disorders are contributing to long-term outcomes for patients, necessitating new approaches to lifelong management.
As patients age, some may begin to manage their own healthcare, making physician-guided transition to the adult setting critical.3 Physicians should ensure the following:
The transition from paediatric to adult care and long-term adult care are critical areas to address in care plans for adolescent and adult patients.3 Long-term care considerations are ideally best addressed in a centre with significant MPS experience, and they require careful coordination across specialities.3,14 Long-term issues include but are not limited to the following:
Long-term management of MPS disorders, including ongoing assessments and a site-specific transition strategy from paediatric to adult care, may lead to sustained improvement in quality of life and a better future for your patients.3,14–16
Because clinical manifestations of mucopolysaccharidosis (MPS) disorders are multisystemic, a patient-specific, multidisciplinary approach is required to proactively recognise and manage complications. The involvement of a cardiologist in this process is key, as cardiac surgeries are frequently performed for patients with MPS.1-4
Patients with MPS disorders typically have a number of surgical interventions over their lifetimes. A natural history study assessing a cohort of 325 patients with Morquio A (MPS IVA) found that over 70% of patients had at least one surgical procedure.5

Patients with MPS have a high perisurgical mortality rate due to multiple factors, including upper and lower airway obstruction, cervical spinal instability, respiratory impairment, cardiovascular morbidities, and frequent infections.3,5,6 For example, surgical complications resulted in an 11% mortality rate in patients with Morquio A (n=27).7
Creating a surgical plan is crucial and involves a multidisciplinary team of specialists who are, ideally, also experienced in treating patients with MPS.6
Cardiac-specific procedural care often includes surgical management of a variety of manifestations. Documented successful cardiac operative procedures in patients with MPS include the following:4
Surgical risk assessment and perioperative monitoring are fundamental components of a tailored surgical plan, and they can reduce the risks of negative surgical outcomes and mortality in patients with MPS.6,10,11


References: 1. McGill JJ, Inwood AC, Coman DJ, et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age – a sibling control study. Clin Genet. 2010;77(5):492–498. doi:10.1111/j.1399-0004.2009.01324.x. 2. Furujo M, Kubo T, Kosuga M, Okuyama T. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol Genet Metab. 2011;104(4):597–602. doi:10.1016/j.ymgme.2011.08.029. 3. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50(suppl 5):v13–18. 4. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 5. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19–v25. doi:10.1093/rheumatology/ker397. 6. Muenzer J, Beck M, Eng CM, et al.Genet Med. 2011;13(2):95–101. doi:10.1097/GIM.0b013e3181fea459. 7. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 8. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63–72. doi:10.1016/j.ymgme.2013.11.015. 9. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. doi:10.1002/ajmg.a.36833. 10. Bagewadi S, Roberts J, Mercer J, Jones S, Stephenson J, Wraith JE. Home treatment with Elaprase® and Naglazyme® is safe in patients with mucopolysaccharidoses types II and VI, respectively. J Inherit Metab Dis. 2008;31(6):733-737. doi:10.1007/s10545-008-0980-0. 11. BioMarin Pharmaceutical Inc. VIMIZIM website. http://www.vimizim.com/. Accessed December 21, 2015. 12. BioMarin Pharmaceutical Inc. NAGLAZYME website. http://www.naglazyme.com/. Accessed December 21, 2015. 13. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19–29. doi:10.1542/peds.2008-0416.
References: 1. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed December 15, 2015. 2. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27–S34. 3. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. doi:10.1002/ajmg.a.36833. 4. Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34(6):1183–1197. doi:10.1007/s10545-011-9359-8. 5. Mohan UR, Hay AA, Cleary MA, Wraith JE, Patel RG. Cardiovascular changes in children with mucopolysaccharide disorders. Acta Paediatr. 2002;91(7):799–804. 6. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19–29. doi:10.1542/peds.2008-0416. 7. Solanki GA, Martin KW, Theroux MC, et al. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome): presentation, diagnosis and management. J Inherit Metab Dis. 2013;36(2):339-355. doi:10.1007/s10545-013-9586-2. 8. Zafeiriou DI, Batzios SP. Brain and spinal MR imaging findings in mucopolysaccharidoses: a review. AJNR Am J Neuroradiol. 2013;34(1):5–13. doi:10.3174/ajnr.A2832. 9. Lachman R, Martin KW, Castro S, Basto MA, Adams A, Teles EL. Radiologic and neuroradiologic findings in the mucopolysaccharidoses. J Pediatr Rehabil Med. 2010;3(2):109–118. doi:10.3233/PRM-2010-0115. 10. Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007;120:405–418. doi:10.1542/peds.2006-2184. 11. Neufeld EF, Muenzer J. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, et al, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001:3421–3452. 12. Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. doi:10.1186/1750-1172-6-72. 13. James A, Hendriksz CJ, Addison O. The oral health needs of children, adolescents and young adults affected by a mucopolysaccharide disorder. JIMD Rep. 2012;2:51–58. doi:10.1007/8904_2011_46. 14. Coutinho MF, Lacerda L, Alves S. Glycosaminoglycan storage disorders: a review. Biochem Res Int. 2012;2012:471325. doi:10.1155/2012/471325. 15. Kakkis ED, Neufeld EF. The mucopolysaccharidoses. In: Berg BO, ed. Principles of child neurology. New York, NY: McGraw-Hill; 1996:1141–1166. 16. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41–v48.
References: 1. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27–S34. 2. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19–29. doi:10.1542/peds.2008-0416. 3. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. doi:10.1002/ajmg.a.36833. 4. Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis. 2011;34(6):1183–1197. doi:10.1007/s10545-011-9359-8. 5. Harmatz P, Mengel KE, Giugliani R, et al. The Morquio A clinical assessment program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109(1):54–61. doi:10.1016/j.ymgme.2013.01.021. 6. Walker R, Belani KG, Braunlin EA, et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211–219. doi:10.1007/s10545-012-9563-1. 7. Lavery C, Hendriksz C. Mortality in patients with Morquio syndrome A. J Inherit Metab Dis Rep. 2015;15:59–66. doi:10.1007/8904_2014_298. 8. Theroux MC, Nerker T, Ditro C, Mackenzie WG. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth. 2012;22(9):901–907. doi:10.1111/j.1460-9592.2012.03904.x. 9. Scarpa M, Almassy Z, Beck M, et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. doi:10.1186/1750-1172-6-72. 10. Solanki GA, Martin KW, Theroux MC, et al. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome): presentation, diagnosis and management. J Inherit Metab Dis. 2013;36(2):339-355. doi:10.1007/s10545-013-9586-2. 11. Vitale MG, Skaggs DL, Pace GI, et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333–339. doi:10.1016/j.jspd.2014.05.003. 12. Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012;107:15–24. doi:10.1016/j.ymgme.2012.07.018. 13. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1–10. doi:10.1155/2013/791983.
