


Kardeşler arasında yapılan çalışmaların destekleyici klinik bulguları, erken müdahalenin hastalığa özgü yönetim ve kullanılabilir olduğu takdirde ERT’nin erken başlatılması yoluyla hastaların sonuçlarını iyileştirmek için birçok fırsat sunduğunu göstermektedir.1-6
Yaşamın erken döneminde veya daha ileriki yaşlarda başlatılan ERT’nin, yaşam kalitesi, ambulasyonun korunması ve günlük yaşam aktiviteleri için hayati önem taşıyan dayanıklılık ve pulmoner önlemler gibi önemli klinik parametreleri iyileştirdiği gösterilmiştir.7,8
ERT şu anda birçok ülkede mukopolisakkaridoz (MPS) I, II, IVA ve VI bulunan hastaların tedavisinde kullanılmaktadır.
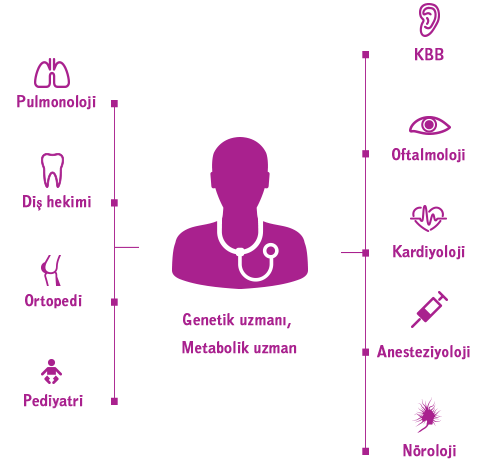
Mukopolisakkaridoz (MPS) hastalıkları gibi progresif, karmaşık, genetik koşullar için yeni yönetim çağı, her bir hastanın sağlık ekibinin etkin koordinasyonuna bağlıdır.1 Genetik uzmanları ve/veya metabolizma uzmanları genellikle evde sağlık hizmetinin merkezinde bulunur ve multidisipliner tedavi ile birlikte bireyselleştirilmiş bir yönetim planının koordinasyonuna yardımcı olurlar.2,3
Birçok MPS hastalığı için, MPS’nin yaşam boyu yönetimi ile ilgili olarak,
kullanılabilir yönetim kılavuz bilgileri ve uzmanlık alanına özgü konsensüs önerileri bulunmaktadır. Kılavuz bilgileri tipik olarak aşağıdakileri önerir:2,4
Koordineli bir tedavi ekibi tarafından yapılan erken ve devam eden değerlendirmeler, hastaların sonuçlarını iyileştirebilir ve geri dönüşü olmayan hasarın önlenmesine yardımcı olabilir.4
Örnek olarak, aşağıdaki tablo Morquio A (MPS IVA) bulunan hastalar için önerilen değerlendirme planını temsil etmektedir.2

Değerlendirmelerin sıklığı ve belirli uzmanların katılımı, farklı MPS tipleri arasında değişiklik gösterir. MPS I, II ve III gibi birincil nörodejeneratif ve bilişsel komplikasyonlarla ilişkili MPS hastalıkları bulunan hastalar için, ilave ve düzenli nörodavranışsal ve psikiyatrik değerlendirmeler önerilir.4-6
MPS’li hastalar için olumlu uzun vadeli sonuçlar alınmasının kolaylaştırılması için yapılması gereken uzmanlık alanına özgü değerlendirmelere ek olarak, genellikle sağlıkla ilgili genetik uzmanı ve/veya metabolizma uzmanı olan koordinatör hekim tarafından önemli adımlar atılabilir. Bunların, diğer sağlık uzmanlarına (örn. diş hekimleri, fizyoterapistler, Pediyatri uzmanları, aile hekimleri) ve ailelere, hastalık ve genel yönetim stratejileri hakkında eğitim vermeye dair rolü son derece önemlidir ve aşağıdakileri içermelidir2:
Uzmanlık alanına özgü değerlendirmelerle birlikte, düzenli fizik muayeneler ve genel sağlık müdahaleleri, MPS alt tipleri arasında değişiklik gösterebilecek, önerilen kılavuzlara uymalıdır.2
MPS hastalıklarının tedavisine dair gelişmeler, hastaların uzun vadeli sonuçlarına katkıda bulunmakta olup, yaşam boyu yönetim için yeni yaklaşımlar gerektirmektedir.
Hastaların yaşı ilerledikçe, bazıları kendi sağlıklarını yönetmeye başlayabilir ve bu da yetişkin duruma hekim rehberliğinde geçiş yapılmasını son derece önemli kılmaktadır.2 Hekimler aşağıdakileri sağlamalıdır:
Pediyatrik tedaviden yetişkin tedavisine ve uzun vadeli yetişkin tedavisine geçiş, ergen ve yetişkin hastalar için tedavi planlarında ele alınması gereken son derece önemli alanlardır.2 Uzun vadeli tedavi hususları, ideal olarak önemli derecede MPS deneyimine sahip bir merkezde en iyi şekilde ele alınır ve tüm uzmanlık alanlarında dikkatli koordinasyona ihtiyaç duyarlar.2,8 Uzun vadeli konulara aşağıdakiler dahildir, ancak bunlarla sınırlı değildir:
MPS hastalıklarının uzun vadeli yönetimi (devam eden değerlendirmeler ve pediyatrik tedaviden yetişkin tedavisine doğru bölgeye özgü bir geçiş stratejisi de dahil olmak üzere), yaşam kalitesinde sürdürülebilir iyileşme ve hastalarınız için daha iyi bir gelecek sağlayabilir.2,8-10
Mukopolisakkaridoz (MPS) hastalıklarının klinik bulguları multisistemik olduğundan dolayı, komplikasyonları proaktif olarak tanımak ve yönetmek için hastaya özgü, multidisipliner bir yaklaşım gereklidir.1 MPS hastalıkları olan hastalar, tipik olarak yaşamları boyunca birçok cerrahi müdahale geçirir. Morquio A (MPS IVA) bulunan 325 hastadan oluşan bir kohortun değerlendirildiği bir doğal seyir çalışmasında, hastaların %70’inden fazlasının en az bir cerrahi prosedür geçirdiği bulunmuştur.2

MPS’li hastalar, üst ve alt solunum yolu obstrüksiyonu, servikal omurilik instabilitesi, solunum yetmezliği, kardiyovasküler morbiditeler ve sık sık meydana gelen enfeksiyonlar gibi çok sayıda faktöre bağlı olarak, yüksek perisürjikal ölüm oranına sahiptirler.2-4 Örneğin, cerrahi komplikasyonlar Morquio A bulunan hastalarda %11 ölüm oranına neden olmuştur (n=27).5
Cerrahi bir plan oluşturmak çok önemlidir ve ideal olarak MPS’li hastaların tedavisinde de deneyimli, multidisipliner bir uzman ekibini içerir.3
Cerrahi risk değerlendirmesi ve perioperatif izleme, özelleştirilmiş bir cerrahi planın temel bileşenleri olup, MPS’li hastalarda olumsuz cerrahi sonuç ve ölüm oranı risklerini azaltabilir.3,9,10


Referanslar: 1. McGill JJ et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age – a sibling control study. Clin Genet. 2010;77(5):492–498. 2. Furujo M et al. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol Genet Metab. 2011;104(4):597–602. 3. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50(suppl 5):v13–18. 4. Lehman TJA et al. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41–v48. 5. Morishita K et al. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19–v25. 6. Muenzer J et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95–101. 7. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91–95. 8. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63–72. 9. Hendriksz CJ et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. 10. Bagewadi S et al. Home treatment with Elaprase® and Naglazyme® is safe in patients with mucopolysaccharidoses types II and VI, respectively. J Inherit Metab Dis. 2008;31(6):733–737. 11. BioMarin Pharmaceutical Inc. VIMIZIM website. http://www.vimizim.com/. Accessed December 21, 2015. 12. BioMarin Pharmaceutical Inc. NAGLAZYME website. http://www.naglazyme.com/. Accessed December 21, 2015. 13. VIMIZIM [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2014. 14. Wood TC et al. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36(2):293–307. 15. NAGLAZYME [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2013. 16. Harmatz P et al. for MPS VI Phase 3 Study Group. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or RHASB) and follow-on, open-label extension study. J Pediatr. 2006;148(4):533–539. 17. Harmatz P et al. for MPS VI Study Group. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab. 2008;94(4):469–475. 18. Harmatz P et al. Enzyme replacement therapy for mucopolysaccharidosis VI: evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Inherit Metab Dis. 2010;33(1):51–60.
Referanslar: 1. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed December 15, 2015. 2. Hendriksz CJet al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. 3. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27–S34. 4. Muenzer J et al. International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19–29. 5. Neufeld EF et al. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. Vol 3. 8th ed. New York: McGraw-Hill; 2002:2465–2494. 6. Scarpa M et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. 7. James A et al. The oral health needs of children, adolescents and young adults affected by a mucopolysaccharide disorder. JIMD Rep. 2012;2:51–58. 8. Coutinho MF et al. Glycosaminoglycan storage disorders: a review. Biochem Res Int. 2012;2012:471325. 9. Kakkis ED et al. The mucopolysaccharidoses. In: Berg BO, ed. Principles of Child Neurology. New York, NY: McGraw-Hill; 1996:1141–1166. 10. Lehman TJA et al. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41–v48.
Referanslar: 1. Wold SM et al. Role of the pediatric otolaryngologist in diagnosis and management of children with mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2010;74(1):27–31. 2. Harmatz P et al. The Morquio A clinical assessment program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109(1):54–61. 3. Walker R et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211–219. 4. Hendriksz CJ et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. 5. Lavery C et al. Mortality in patients with Morquio syndrome A. J Inherit Metab Dis Rep. 2015;15:59-66. 6. Theroux MC et al. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth. 2012;22(9):901–907. 7. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27–S34. 8. Scarpa M et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. 9. Solanki GA et al. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome): presentation, diagnosis and management. J Inherit Metab Dis. 2013;36(2):339–355. 10. Vitale MG et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333–339. 11. Solanki GAet al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012;107:15-24. 12. Spinello CM et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1–10.
