Onlarca yıldır süren araştırmalar ve klinik deneyim, mukopolisakkaridoz (MPS) hastalıklarının en uygun yönetiminde yeni bir çağ açmıştır. MPS için hızla gelişen bu standart bakım, koordineli, multidisipliner bir bakımı bünyesinde barındıran ve hekimlere hastaların yaşamlarını değiştirmek için eşsiz fırsatlar sağlayan bir sağlık hizmeti sunma modelinin merkezindeki metabolizma veya genetik uzmanına dayanmaktadır.1–3
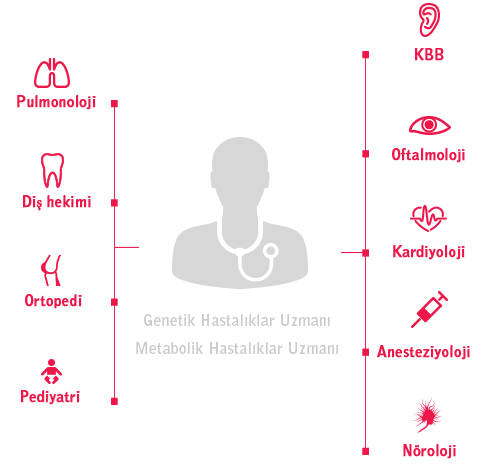
MPS hastalıklarının heterojen ve değişken doğası, koordineli hasta bakımı için kişiselleştirilmiş bir yaklaşım gerektirmektedir.4 Koordineli bakımın amacı, aşağıdakiler de dahil olmak üzere hastaların daha yüksek bir yaşam kalitesini elde etmelerine yardımcı olmaktır:
MPS gibi kronik, karmaşık, multisistemik genetik hastalığı olan pediyatrik hastalar için koordineli bir yaklaşım aracılığıyla bakım, sağlık hizmetlerinin kullanımının azaltılması ve sağlık sonuçlarının iyileştirilmesi ile ilişkilidir.5-8
Koordinasyon, geniş sağlık sisteminin tüm unsurları (örn. özel bakım, hastaneler, evde sağlık hizmeti ve toplum hizmetleri) ve hastaların bireyselleştirilmiş yönetim planları dahilinde uygulanmalıdır.3 Rolünüz, MPS hastalıklarının yönetimi ve hasta sonuçlarının iyileştirilmesine dair en iyi uygulamaların tatbik edilmesi açısından hayati olabilir.
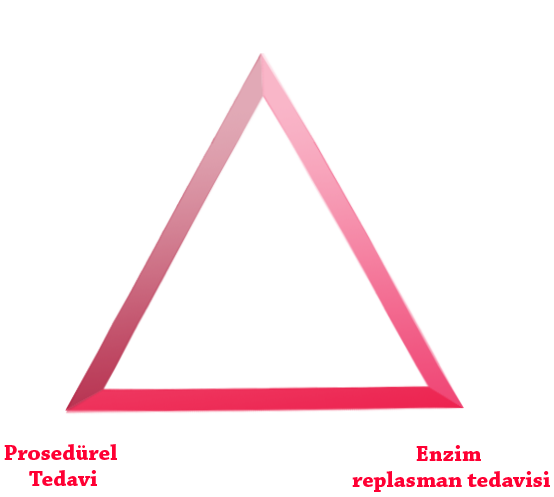
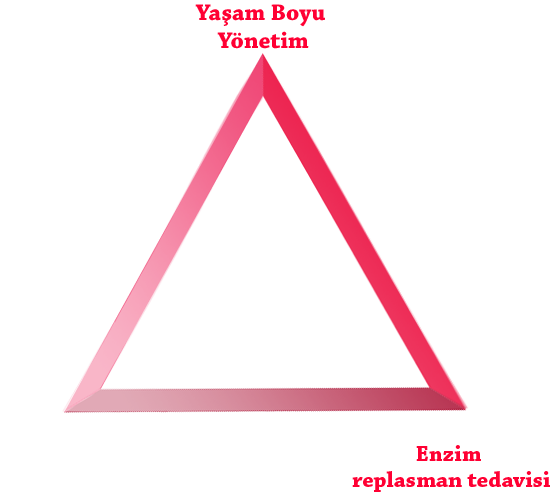
Aşağıda 3 tedavi desteği olarak gruplanmış en uygun MPS hastalığı yönetiminin uygulanması, hasta sonuçlarını iyileştirmeye yardımcı olabilir:
Göz hekimleri, koordineli bir yaklaşımın bir parçası olarak kronik bakımı ve prosedürel bakımı ele alan, bireyselleştirilmiş yönetim planlarına katkıda bulunmakta ve sonuç olarak hasta sonuçlarının en iyi duruma getirilmesine yardımcı olmaktadır.2,10
Yapılabiliyorsa ERT, tedavinin temel taşıdır.9-11
MPS hastalığının her bir alt tipi klinik açıdan birbirinden farklı olsa da, hepsi MPS hastalığının patolojisinde yaygın olarak görülen, yaşamı sınırlayan, progresif, multisistemik hastalık bulguları ortaya koymaktadır.11,13,21,22 MPS'li hastaların yönetiminde, her bir MPS alt tipi için belirli klinik bulguların ve yönetim tavsiyelerinin anlaşılması gerekmektedir.2,10
I
II
III(A, B, C ve D)
α-N-asetilglukozaminidaz, Asetil CoA:
α-glukozaminidN-asetiltransferaz, N-asetilglukozamin-6-sülfataz
IV
β-galaktosidaz
VI(A ve B)
VII(A ve B)
IX
Kasım 2015
Birçok düzenlemeye rağmen, en uygun bilateral doğum sancısı analjezisi sağlanamamış ve sezaryen doğum için sistemik analjeziye ihtiyaç duyulmuştur.
Mayıs 2015
Tanı tesislerinin mevcut olmaması veya uzak olması, alternatif tanılar ve yaşanan yanıltıcı semptomlar nedeniyle, tanıda gecikmeler meydana gelmiştir. Birçok hasta beklenenden daha gizli olan ve bunu takiben göz ardı edilen bulgular yaşamıştır. Vakalar ayrıca, MPS VI'nın farklı uzmanlık dalları açısından tanılanmasıyla ilişkili benzersiz zorlukları vurgulamıştır ve bu hastalarda ilk belirtilerin nasıl görüldüğü konusunda fikirler vermektedir.
Nisan 2016
Özellikle koroner dolaşım ve miyokard sorunu bulunan MPS'li yetişkinlerdeki diğer kalp sorunlarına dair mevcut anlayışımız yetersizdir ve bu öne çıkan yetişkin popülasyonuna etkili şekilde bakım sunmak için daha fazla bilgiye ihtiyaç vardır.
Referanslar: 1. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. doi:10.1002/ajmg.a.36833. 2. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27–S34. 3. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed December 15, 2015. 4. Hendriksz CJ, Harmatz P, Beck M, et al. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol Genet Metab. 2013;110:54–64. doi:10.1016/j.ymgme.2013.04.002. 5. Casey PH, Lyle RE, Bird TM, et al. Effect of hospital-based comprehensive care clinic on health costs for Medicaid-insured medically complex children. Arch Pediatr Adolesc Med. 2011;165(5):392–398. doi:10.1001/archpediatrics.2011.5. 6. Mosquera RA, Avritscher EBC, Samuels CL, et al. Effect of an enhanced medical home on serious illness and cost of care among high-risk children with chronic illness: a randomized clinical trial. JAMA. 2014;312(24):2640–2648. doi:10.1001/jama.2014.16419. 7. Klitzner TS, Rabbitt LA, Chang RKR. Benefits of care coordination for children with complex disease: a pilot medical home project in a resident teaching clinic. J Pediatr. 2010;156(6):1006–1010. doi:10.1016/j.jpeds.2009.12.012. 8. Gordon JB, Colby HH, Bartelt T, Jablonski D, Krauthoefer ML, Havens P. A tertiary care-primary care partnership model for medically complex and fragile children and youth with special health care needs. Arch Pediatr Adolesc Med. 2007;161(10):937–944. 9. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91–95. 10. Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19–29. doi:10.1542/peds.2008-0416. 11. Muenzer J, Beck M, Eng CM, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011;13(2):95–101. doi:10.1097/GIM.0b013e3181fea459. 12. Kakkis ED, Neufeld EF. The mucopolysaccharidoses. In: Berg BO, ed. Principles of child neurology. New York, NY: McGraw-Hill; 1996:1141–1166. 13. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41–v48. 14. Lavery C, Hendriksz C. Mortality in patients with Morquio syndrome A. J Inherit Metab Dis Rep. 2015;15:59–66. doi:10.1007/8904_2014_298. 15. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome) –10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953–1964. doi:10.1002/ajmg.a.36584. 16. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1–10. doi:10.1155/2013/791983. 17. Sam JA, Baluch AR, Niaz RS, Lonadier L, Kaye AD. Mucopolysaccharidoses: anesthetic considerations and clinical manifestations. Middle East J Anaesthesiol. 2011;21(2):243–253. 18. Data on file. Biomarin Pharmaceutical, Inc. 19. Drummond JC, Krane EJ, Tomatsu S, Theroux MC, Lee RR. Paraplegia after epidural-general anesthesia in a Morquio patient with moderate thoracic spinal stenosis. Can J Anesth. 2015;62(1):45–49. doi:10.1007/s12630-014-0247-1. 20. Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schöls L. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78. doi:10.1186/1752-1947-8-78. 21. Clarke LA, Winchester B, Giugliani R, Tylki-Szymańska A, Amartino H. Biomarkers for the mucopolysaccharidoses: discovery and clinical utility. Mol Genet Metab. 2012;106(4):396–402. doi:10.1016/j.ymgme.2012.05.003. 22. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19–v25. doi:10.1093/rheumatology/ker397.