


Mukopolisakkaridoz (MPS) VI veya Maroteaux-Lamy sendromu, birden fazla organı etkileyen, şiddetli patolojilere sahip, yıkıcı, progresif ve heterojen bir hastalıktır. Glikozaminoglikanlar (GAG'ler) olan dermatan sülfat ve kondroitin sülfatı katabolize eden arilsülfataz B (ASB) enziminin yetersiz faaliyet göstermesi nedeniyle ortaya çıkar.1,2
MPS VI'nın fenotipik ortaya çıkışı çok çeşitli şekillerde olabilse de,1,2 hastalar çoğunlukla hızlı ya da yavaş ilerleyen hastalık geçirmeleriyle ayırt edilirler:
İlerleme hızı ne olursa olsun, tedavi edilmeyen MPS VI yıllar içinde ilerleyebilir ve şunlara yol açabilir:3
MPS VI klinik açıdan heterojen bir durum olup, hastalarda belirgin hastalık yaşamın ilk yıllarında görülebilir veya daha yavaş ilerleyen hastalıkta, semptomlar daha uzun süreye yayılmış şekilde ortaya çıkar. Ancak, MPS VI'nın sürekli olarak semptomlar sergilediğini bilmek önemlidir, dolayısıyla bu hastalık ilerleme kategorileri için sabit parametreler bulunmamaktadır.1,2
Hızlı ilerleyen MPS VI, Swiedler ve arkadaşlarının kesitsel anket çalışmasında karakterize edilmiş olup, idrarda 200 μg/mg'den yüksek GAG proteini (uGAG) seviyelerinin,3 spesifik olmayan semptomlarla birlikte yaşamın ilk yıllarında kendini gösterebildiğini öne sürmektedir. 2. ve 3. yıllar arasında, aşağıdakiler de dahil olmak üzere karakteristik özellikler ortaya çıkar:1,2,5
uGAG seviyeleri 200 μg/mg'den daha az veya 200 μg/mg'ye eşit olarak karakterize edilen, yavaş ilerleyen MPS VI, açık bir şekilde ortaya çıkmayabilir ve bu nedenle tanıyı yaşamın geç evrelerine kadar geciktirebilir. Bununla birlikte, hastalar yaşamı kısıtlayabilecek ve tehdit edebilecek derecede önemli morbidite sergilerler. Fiziksel kısıtlamalar hastaların öğrenim ve gelişimini etkileyebilse de, hızlı veya yavaş ilerleyen MPS VI genellikle nörobilişsel yetersizliklere neden olmaz.3 Aşağıdaki tablo, organ sistemine göre MPS VI'nın komplikasyonlarını özetlemektedir. MPS VI ile ilişkili klinik bulgular heterojendir; ancak, tüm hastalarda hastalığın ilerlediği görülür.

MPS VI bulunan hastalar, etkilenmeyen, yaşa göre düzeltilmiş akranlarına göre belirgin derecede daha düşük büyüme hızları sergilerler. MPS VI bulunan hastaların yaşadığı büyüme sapması, ikincil sorunların bir işareti olabilir ve kötü beslenme, endokrin anomalileri veya psikososyal yoksunluk gibi sorunlar için klinik gözlem yapılmasını gerektirir.

MPS VI, başlangıçları ve ilerleme hızları değişiklik gösteren bir fenotip spektrumuna sahip, klinik açıdan heterojen olan bir durumdur.2 Aşağıdaki şekil, hastalığı hızlı ilerleyen bir hasta ile yavaş ilerleyen bir hasta arasında geniş çapta görülen değişkenliği ortaya koymaktadır. Hastalığı hızlı ilerleyen hasta, MPS VI'nın fenotipik ayırıcı özelliklerinin birçoğunu sergiler.
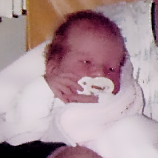
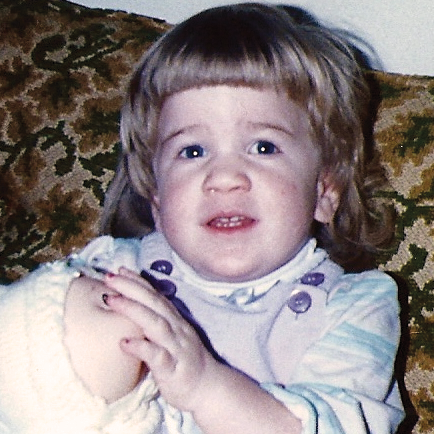


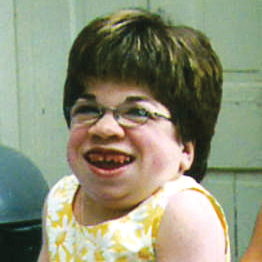
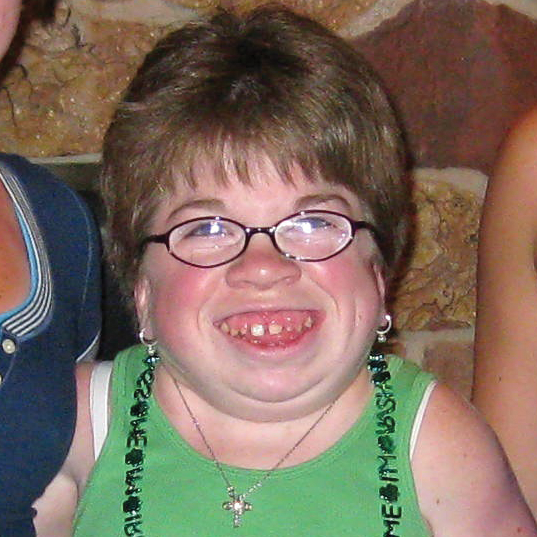
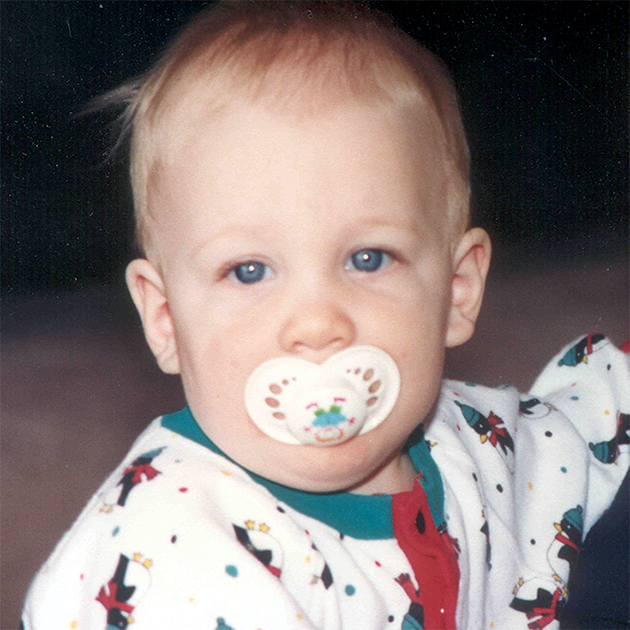

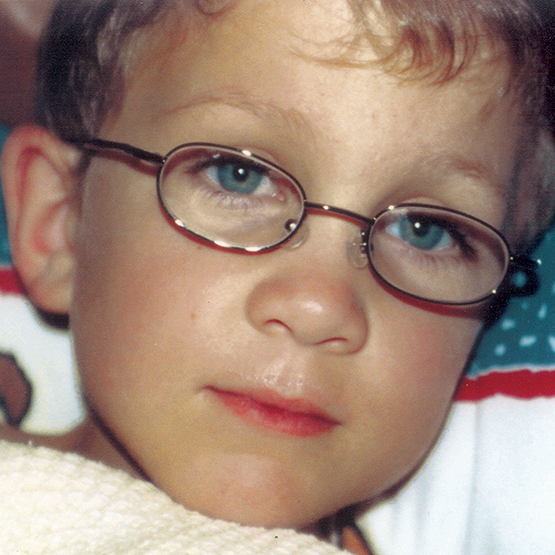



Hızlı ve yavaş ilerleyen hastalığa farklılaşma, sadece aşırı durumlarda belirgin olur. Tedavi edilmezse, hastalığın ilerlemesi fiziksel ve fonksiyonel sağlığın iyi olmasını engeller ve sonuç olarak ömrü belirgin derecede kısaltır.4
Fenotip ne olursa olsun, etkilenen bireylerin hastalığı yıllar geçtikçe ilerler ve sonuç olarak şunları yaşarlar:4
MPS VI bulunan hastalar, genellikle enfeksiyonlar, cerrahiye bağlı komplikasyonlar veya kardiyopulmoner hastalık nedeniyle ölür.4
MPS VI, N-asetilgalaktozamin 4-sülfataz (ASB olarak da bilinir) enziminin eksikliğinden kaynaklanan, otozomal, resesif bir durumdur. Bu eksiklik, dermatan sülfat GAG'inin çeşitli dokuların lizozomlarında birikmesine neden olur.13,14

Yüksek MPS VI riski ile hiçbir belirli etnik grup ilişkilendirilmemiştir; ancak, belirli mutasyonların sıklığının arttığı bazı durumlar bildirilmiştir. MPS VI bulunan Brezilyalı hastalarda, alellerin %23'ünde ortak bir mutasyon (1533del23) bulunmuştur. Buna ek olarak, bir çalışma Almanya'da yaşayan Türk popülasyonunda, Türk olmayan Alman popülasyonuna göre MPS VI'nın yüksek doğum prevalansı sergilediğini ortaya koymuştur.2
Geçtiğimiz on yılda MPS VI'nın yönetimi, klinisyenlerin hastalığa dair fenotipik değişkenlik ve ilerleme gibi konularda bilgi sahibi olması ile birlikte gelişmiştir. Yetişkinlik dönemine giren hastalar gibi diğer faktörler, yönetime ilişkin araştırma, anlayış ve potansiyel için yeni alanları vurgulamaktadır.
MPS VI birden fazla vücut sistemini etkiler ve çok çeşitli hastalık bulgularının yönetimini, entegre tedavinin önemli bir parçası haline getirir. Yönetim, uyarlamalı veya destekleyici cihazların kullanımını, fiziksel ve mesleki terapiyi, semptom bazlı ilaçları, cerrahi müdahaleleri ve eksik enzimi tedarik etmeye yönelik tedaviyi içermelidir.4
2007 yılında yayınlanan "Mukopolisakkaridoz İçin Yönetim Kılavuzları", MPS VI hastalarının yönetimi için tüm uzmanlık alanlarına yönelik tavsiyeler sunmaktadır. MPS VI bulunan hastalarda düşünülmesi gereken bir tedavi seçeneği olarak, enzim replasman tedavisini (ERT) önermektedir.4 Kılavuzlar, uygun olan durumlarda ERT'nin başlatılmasının yanı sıra, GAG birikimi ile ilişkili semptomlar için sisteme özgü yönetim stratejileri önermektedir.4 Multidisipliner, koordineli bir tedavi ekibinin yürüttüğü yaşam boyu yönetim ve prosedürel bakım, hastaların sonuçlarını en uygun duruma getirmenin son derece önemli bileşenleridir.14,15 MPS VI bulunan hastalar, birden fazla organ sisteminde hastalığın ilerlemesinin değerlendirilmesi ve hastalığın potansiyel komplikasyonlarının tespiti ve tedavisi için, erken ve düzenli değerlendirmelere ihtiyaç duyarlar.4 Aşağıdaki tablo, MPS VI bulunan hastalar için 2007 kılavuzları tarafından önerilen değerlendirme planını göstermektedir.

MPS VI bulunan hastalar, çoğunlukla hastalığın multisistemik komplikasyonlarını ele alan cerrahi müdahalelere ihtiyaç duyarlar. Bu cerrahi tedavi, hastalığın doğası gereği karmaşıktır.15
MPS VI bulunan hastalar, cerrahi riski ve izlem ihtiyacını önemli ölçüde artırabilen, aşağıdakilerin de dahil olduğu birçok faktörden dolayı sıkıntı çekerler:15
Bu faktörler, cerrahi ve anestezik tedaviyi zorlaştırır, önceden planlama gerektirir ve en uygun sonuçları artırmak için, hastalığa özgü teknikleri gerekli kılar.16
Entübasyon ve ekstübasyon gibi anestezi sırasında kullanılan özel perioperatif prosedürler ve bir intraoperatif nöroizleme kontrol listesinin kullanımı, başarılı cerrahi müdahaleler için esastır. MPS VI uzmanlarından oluşan entegre bir cerrahi ekip, olumlu, sürekli sonuçlar için son derece önemlidir.15,16
MPS VI bulunan hastalar yetişkinlik dönemine ulaştığında, tıbbi ekipleri ile ilişkileri değişecektir. Bu geçişin yönetimine yardımcı olmak amacıyla, tedavi kesintilerini en aza indirmek, desteği pediyatrik tedavi ve ebeveyn desteğinin kapsamı dışına genişletmek ve yetişkin hastaların MPS VI yönetimine dair bilgili olmasını sağlamak için bireysel planlar gereklidir.17 Bu geçiş planları her bir hastanın belirli ihtiyaçlarına göre özelleştirilmelidir; bu şekilde, kendi bakımlarını üstlenebilecek hastalar gerekli araçlara sahip olacak ve daha kısıtlı olan hastalar, kendilerini desteklemeye dair uygun tedaviye ve hizmetlere sahip olacaktır. Bu planlar, öngörülen hedefine ulaşması için hastanın kapasitesiyle birlikte, bilgisi ve durumu hakkındaki bilgileri iletebilme yeteneğini belirleyen bir değerlendirme içermelidir.17
Referanslar: 1. Thümler A et al. Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: a case series. J Inherit Metab Dis 2012;35(6):1071-1079. 2. Valayannopoulos V et al. Mucopolysaccharidosis VI. Orphanet J Rare Dis 2010;5:5. . 3. Swiedler SJ et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with mucopolysaccharidosis VI (Maroteaux–Lamy syndrome). Am J Med Genet A 2005;134A(2):144-150. 4. Giugliani R et al. Management guidelines for mucopolysaccharidosis VI. Pediatrics 2007;120:405-418. 5. Muhlebach MS et al. Respiratory manifestations in mucopolysaccharidoses. Paediatr Respir Rev 2011;12(2):133-138. 6. Lin H-Y et al. Polysomnographic characteristics in patients with mucopolysaccharidoses. Pediatr Pulmonol. 2010;45(12):1205-1212. 7. Kampmann Cet al. Mucopolysaccharidosis VI: cardiac involvement and the impact of enzyme replacement therapy. J Inherit Metab Dis 2014;37(2):269-276. 8. Willoughby CE et al. Anatomy and physiology of the human eye: effects of mucopolysaccharidoses disease on structure and function — a review. Clin Exper Ophthalmol. 2010;38(suppl 1):2-11. 9. Ganesh A et al. An update on ocular involvement in mucopolysaccharidoses. Curr Opin Ophthalmol 2013;24(5):379-388. 10. Kantaputra PN et al. Oral manifestations of 17 patients affected with mucopolysaccharidosis type VI. J Inherit Metab Dis 2014;37(2):263-268. 11.National Institute of Neurological Disorders and Stroke Mucopolysaccharidoses Fact Sheet. http://www.ninds.nih.gov/disorders/mucopolysaccharidoses/detail_mucopolysaccharidoses.htm. Accessed November 2016. 12. Data on file. BioMarin Pharmaceutical Inc. 13. NAGLAZYME [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2013. 14. Giugliani R et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome) — 10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A 2014;164A(8):1953-1964. 15. Walker R et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis 2013;36(2):211-219. 16. Vitale MG et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333-339. 17. American Academy of Pediatrics, American Academy of Family Physicians, American College of Physicians, Transitions Clinical Report Authoring Group, Cooley WC, Sagerman PJ. Supporting the health care transition from adolescence to adulthood in the medical home. Pediatrics 2011;128(1):182-200.
