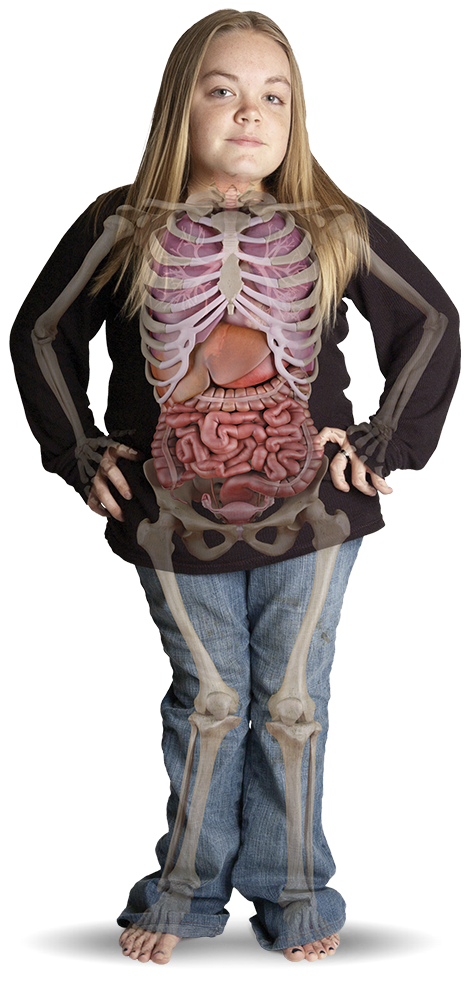
Departmanınızda nadir görülen bir genetik hastalık gözden kaçıyor olabilir mi?
Çoğunlukla sadece bir çocukluk dönemi bozukluğu veya kas-iskelet sistemi hastalığı olarak yanlış tanımlanan MPS, düşündüğünüz kadar seyrek olmayabilir
Mukopolisakkaridoz (MPS) hastalıkları, dünya çapında 22.500 canlı doğumdan 1'ini etkilediğinden şüphelenilen, heterojen ortaya çıkış ve değişken hastalık ilerlemesi gösteren bir kalıtsal enzim eksikliği grubudur.1,2
MPS hastalıkları arasında, benzer klinik bulgular ve ilerleme hızları görülmektedir. Aşağıdaki resimler, MPS VI geçiren bir hastada görülen şekliyle MPS hastalığının ilerleme şekillerini göstermektedir.
MPS VI bulunan hastalarda değişken hastalık ilerlemesi
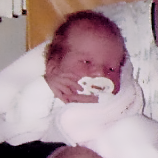
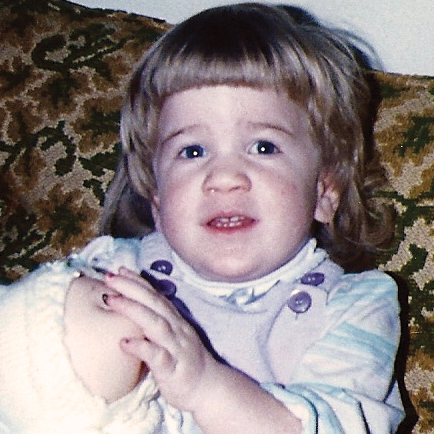


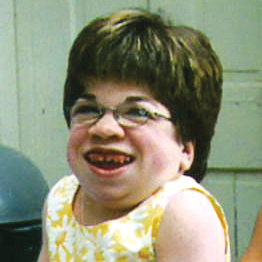
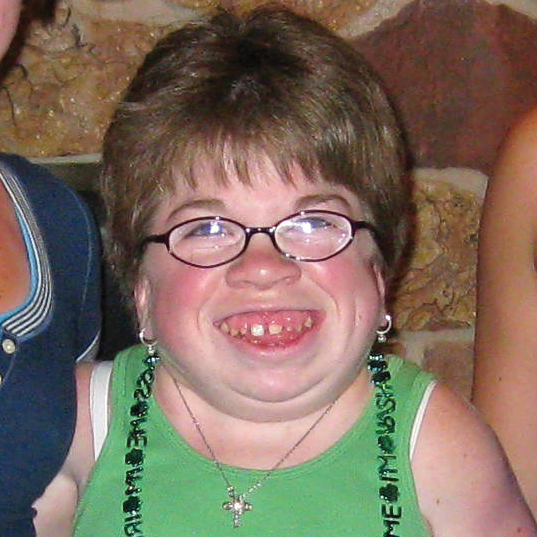
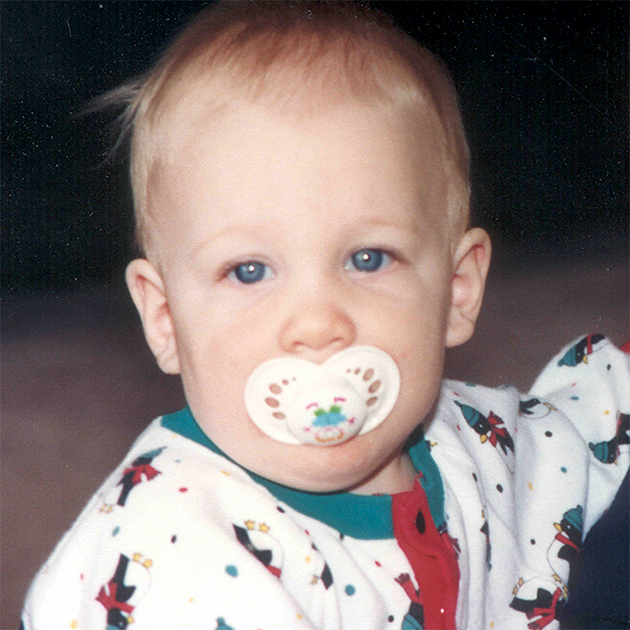

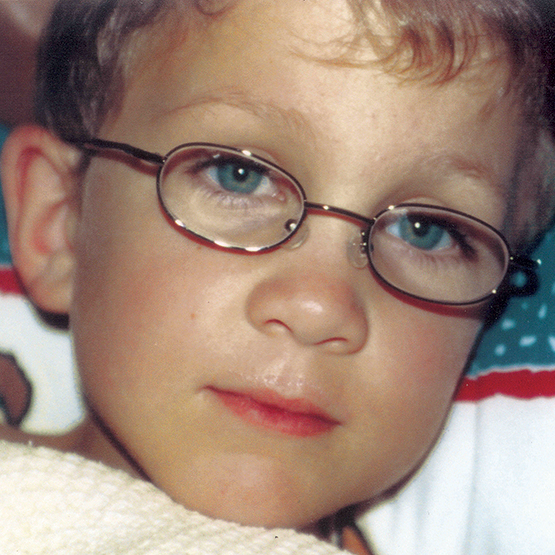



Tanı sık sık geç konulur ve bu hastalar için yıkıcı sonuçlar doğurabilir3
Hastalığın nadir görülen olarak algılanması, hastalığın ortaya çıkışı ve ilerleyişindeki değişkenlik ve MPS ile ilişkili sayısız spesifik olmayan semptom, tanıyı zorlaştırmaktadır. İlk semptomun görülmesinden tanıya kadar geçen zaman, altı aydan on yıllara kadar değişiklik gösterebilir.3

Erken tanı, hasta sonuçlarını en iyi duruma getirmek için son derece önemlidir3,5
Erken tanı, hastalığa özgü yönetime erişim ve enzim replasman tedavisi (ERT) aracılığıyla hasta sonuçlarını iyileştirebilir.3,5–8 Birçok MPS hastalığı için, halihazırda kullanılan veya geliştirilmekte olan bir ERT mevcuttur. MPS'yi tanılamanın ve bunu takiben tedaviyi başlatmanın en iyi yolu, şüphelendiğiniz hastaları bunun testi hakkında bilgi sahibi olan bir genetik uzmanına veya metabolizma uzmanına sevk etmektir.9
MPS'nin erken belirtilerini ve semptomlarını arayın, özellikle:3,10–12
- Klasik
- Fıtıklar
- Eklem ve iskelet anormallikleri
- Boy kısalığı
- Fiziksel görünümde her türlü kademeli, progresif değişiklikler
- Klasik olmayan
- Kalınlaşmış kaburga veya köprücük kemikleri, hafif çarpık bacaklar ve eklem anomalileri veya ağrısı dahil olmak üzere, hemen göze çarpmayan iskelet anomalileri belirtileri
- Egzersiz kapasitesinde azalma ve/veya açıklanamayan kardiyovasküler tutulum, özellikle pediyatrik hastalarda kalp hırıltısı
- Açıklanamayan işitme veya görme bozukluğu (korneada bulanıklaşma)