


Клинические данные на основе исследований сибсов подтверждают, что своевременное начало лечения ведет к формированию разнообразных возможностей улучшения результатов лечения пациентов за счет соответствующей тактики лечения и начала ФЗТ – по возможности в кратчайшие сроки.1–6
Было показано, что начало ФЗТ на ранних или более поздних этапах в жизни улучшает основные клинические параметры, такие как выносливость и показатели функции легких, которые имеют решающее значение для качества жизни, поддержания способности к передвижению и повседневной жизни.7,8
В настоящее время ФЗТ доступна во многих странах для лечения пациентов с МПС I, II, IVA и VI типов
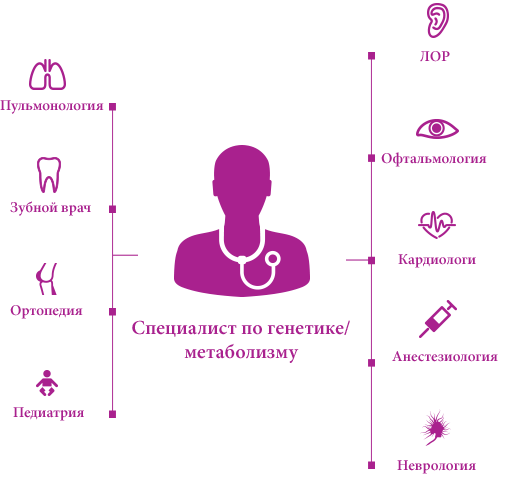
Новая эпоха лечения прогрессирующих комплексных генетических нарушений, например мукополисахаридоза (МПС), зависит от эффективной координации деятельности каждой группы врачей, работающих с пациентом.1
Обычно в медицинском центре есть генетики и/или специалисты по нарушению обмена веществ – они помогают скоординировать работу многопрофильной группы врачей и составить индивидуальный план лечения.2,3
Для МПС разных типов разработаны протоколы ведения больных и
специализированные усредненные рекомендации относительно непрерывного лечения МПС. Обычно рекомендуются к проведению следующие мероприятия:2,4
Своевременно и регулярно проводимые группой специалистов оценки состояния пациента могут улучшить результаты лечения и содействовать профилактике необратимого ущерба.4
В приведенных ниже таблицах представлены примерные графики обследований пациентов с синдромом Моркио типа A (МПС IVA).2
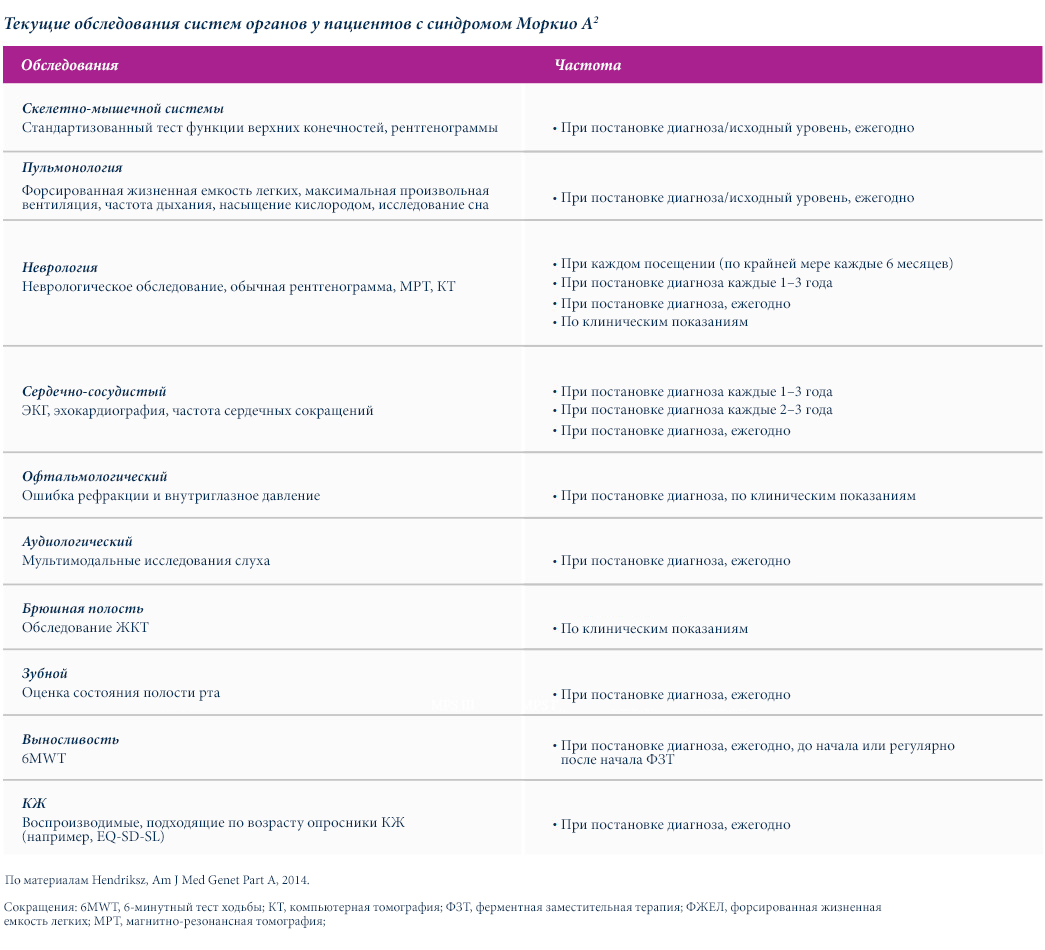
Частота проведения обследований и участие конкретных специалистов различаются в зависимости от типа МПС. Для пациентов с МПС с первичными нейродегенеративными и когнитивными осложнениями (например, МПС I, II и III типов) рекомендуются дополнительные и регулярные нейробиологические и психиатрические обследования.4–6
В дополнение к специализированным обследованиям, которые должны проводиться с целью обеспечения положительных долгосрочных результатов пациентов с МПС, ведущий врач (как правило, генетик и/или специалист по нарушениям обмена веществ) может предпринять важные меры, связанные с общим состоянием здоровья. Функции этих специалистов в информировании врачей других специальностей (например, стоматологов, физиотерапевтов, педиатров, семейных врачей) и членов семей о специфике заболевания и общих стратегиях лечения имеют решающее значение и должны включать2:
Специализированные обследования, а также регулярные медицинские осмотры и общие медико-санитарные вмешательства должны следовать установленным рекомендациям, которые могут варьироваться в зависимости от типов МПС.2
Совершенствование методов лечения МПС способствуют улучшению долгосрочных результатов пациентов, поэтому важно разрабатывать новые подходы к лечению на протяжении всей жизни.
По мере взросления пациентов некоторые из них могут приступить к самостоятельному контролю своего здоровья. Следовательно, важное значение приобретает переход к взрослому этапу жизни при помощи лечащего врача.2 Врачи должны обеспечить следующие условия:
Переход от ведения группой врачей-педиатров к лечению специалистами в области терапии взрослых пациентов и долгосрочное лечение взрослых пациентов – важнейшие элементы, которые необходимо учесть при составлении индивидуальных планов лечения подростков и взрослых.2 Оптимальная реализация долгосрочной терапии наиболее возможна в медицинском центре, где специалисты обладают значительным опытом работы с МПС, притом что требуется продуманная координация действий врачей разных специальностей.2,8 Вопросы долгосрочной терапии включают следующие аспекты (но не ограничиваются ими):
Следствием долгосрочного ведения пациентов с МПС, в том числе непрерывных обследований и стратегии перехода от педиатрии к медицинской помощи взрослым, могут стать устойчивое повышение качества жизни и более благоприятное будущее ваших пациентов.2,8–10
Поскольку клинические проявления мукополисахаридозов являются полиорганными, необходимо применять многопрофильный индивидуальный подход к пациенту, чтобы проактивно выявлять и проводить лечение осложнений.1
Пациентам с МПС в течение жизни обычно проводится несколько хирургических вмешательств. Исследование естественного развития заболевания, в котором оценивалась когорта из 325 пациентов с синдромом Моркио типа A (МПС IVA), показало, что у более 70% пациентов состоялось по крайней мере одно хирургическое вмешательство.2
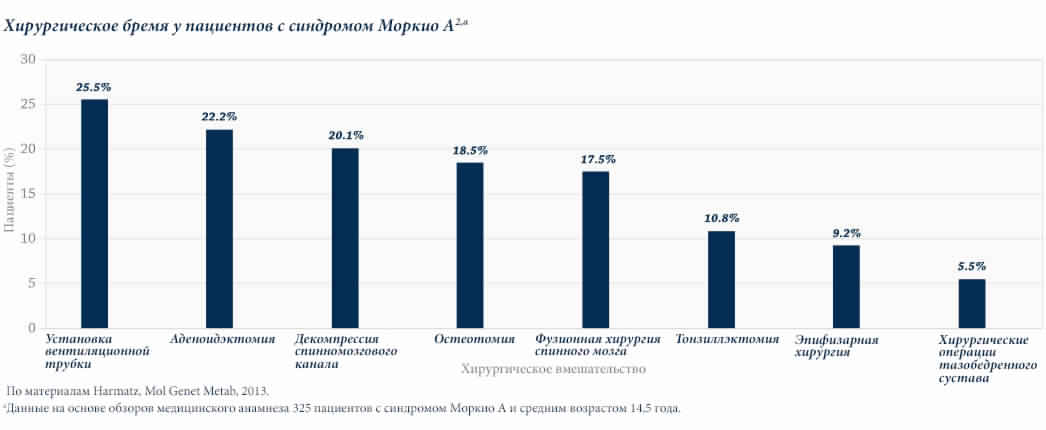
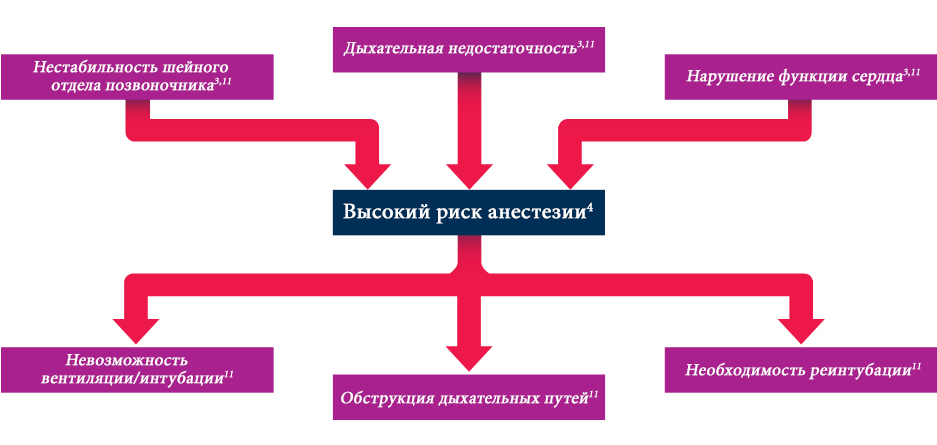
Для пациентов с МПС характерны высокие показатели периоперационной смертности по причине множественных факторов, включая обструкцию верхних и нижних дыхательных путей, нестабильность шейного отдела позвоночника, нарушение дыхания, сердечно-сосудистую патологию и частые инфекции.2–4 Так, в результате хирургических осложнений смертность пациентов с синдромом Моркио типа A составила 11% (n = 27).5
Решающее значение имеет формирование плана хирургических вмешательств, включающее работу мультидисциплинарной группы специалистов, которые в идеале также имеют опыт лечения пациентов с МПС.3
Оценка риска хирургических вмешательств и периоперационный мониторинг являются фундаментальными компонентами индивидуального хирургического плана. Кроме того, они могут снизить риск отрицательных хирургических результатов и смертности у пациентов с МПС.3,9,10

References: 1. McGill JJ et al. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age – a sibling control study. Clin Genet. 2010;77(5):492–498. 2. Furujo M et al. Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Mol Genet Metab. 2011;104(4):597–602. 3. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50(suppl 5):v13–18. 4. Lehman TJA et al. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41–v48. 5. Morishita K et al. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19–v25. 6. Muenzer J, Beck M, Eng CM, et al.Genet Med. 2011;13(2):95–101. doi:10.1097/GIM.0b013e3181fea459. 7. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91–95. 8. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63–72. 9. Hendriksz CJ et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. 10. Bagewadi S et al. Home treatment with Elaprase® and Naglazyme® is safe in patients with mucopolysaccharidoses types II and VI, respectively. J Inherit Metab Dis. 2008;31(6):733–737. 11. BioMarin Pharmaceutical Inc. VIMIZIM website. http://www.vimizim.com/. Accessed December 21, 2015. 12. BioMarin Pharmaceutical Inc. NAGLAZYME website. http://www.naglazyme.com/. Accessed December 21, 2015. 13. VIMIZIM [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2014. 14. Wood TC et al. Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis. 2013;36(2):293–307. 15. NAGLAZYME [package insert]. Novato, CA: BioMarin Pharmaceutical Inc; 2013. 16. Harmatz P et al. for MPS VI Phase 3 Study Group. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or RHASB) and follow-on, open-label extension study. J Pediatr. 2006;148(4):533–539. 17. Harmatz P et al. for MPS VI Study Group. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab. 2008;94(4):469–475. 18. Harmatz P et al. Enzyme replacement therapy for mucopolysaccharidosis VI: evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Inherit Metab Dis. 2010;33(1):51–60.
References: 1. Agency for Healthcare Research and Quality. Defining the PCMH. https://pcmh.ahrq.gov/page/defining-pcmh. Accessed December 15, 2015. 2. Hendriksz CJet al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. 3. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27–S34. 4. Muenzer J et al. International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19–29. 5. Neufeld EF et al. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. Vol 3. 8th ed. New York: McGraw-Hill; 2002:2465–2494. 6. Scarpa M et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. 7. James A et al. The oral health needs of children, adolescents and young adults affected by a mucopolysaccharide disorder. JIMD Rep. 2012;2:51–58. 8. Coutinho MF et al. Glycosaminoglycan storage disorders: a review. Biochem Res Int. 2012;2012:471325. 9. Kakkis ED et al. The mucopolysaccharidoses. In: Berg BO, ed. Principles of Child Neurology. New York, NY: McGraw-Hill; 1996:1141–1166. 10. Lehman TJA et al. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41–v48.
References: 1. Wold SM et al. Role of the pediatric otolaryngologist in diagnosis and management of children with mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2010;74(1):27–31. 2. Harmatz P et al. The Morquio A clinical assessment program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109(1):54–61. 3. Walker R et al. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36(2):211–219. 4. Hendriksz CJ et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1–15. 5. Lavery C et al. Mortality in patients with Morquio syndrome A. J Inherit Metab Dis Rep. 2015;15:59-66. 6. Theroux MC et al. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth. 2012;22(9):901–907. 7. Muenzer J. The mucopolysaccharidoses: a heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004;144(suppl 5):S27–S34. 8. Scarpa M et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis. 2011;6:72. 9. Solanki GA et al. Spinal involvement in mucopolysaccharidosis IVA (Morquio-Brailsford or Morquio A syndrome): presentation, diagnosis and management. J Inherit Metab Dis. 2013;36(2):339–355. 10. Vitale MG et al. Delphi Consensus Report: Best practices in intraoperative neuromonitoring in spine deformity surgery: development of an intraoperative checklist to optimize response. Spine Deformity. 2014;2(5):333–339. 11. Solanki GAet al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012;107:15-24. 12. Spinello CM et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1–10.
