Может ли в вашем отделении находиться пациент с редким генетическим заболеванием?
МПС часто ошибочно определяют как заболевание исключительно детского возраста или заболевание опорно-двигательного аппарата, но оно может быть не настолько редким, как вы думаете
Мукополисахаридозы (МПС) представляют собой группу наследственных заболеваний, вызываемых ферментной недостаточностью и различающихся по своим клиническим проявлениям и характеру прогрессирования. Согласно исследованиям,предполагаемаяраспространенность этих заболеваний в мире составляет 1 на 22 500 новорожденных.1,2
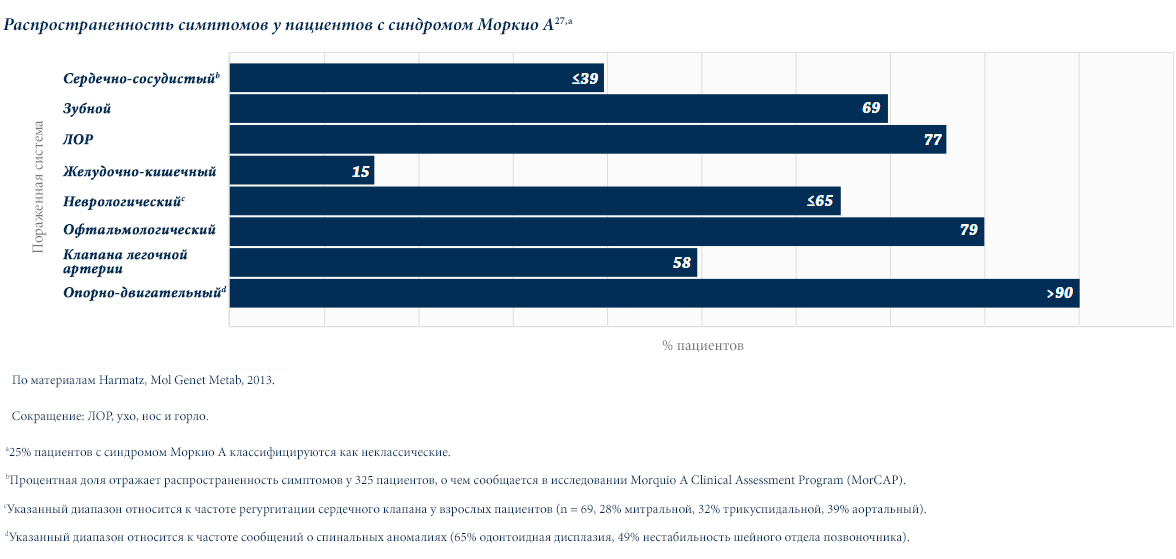
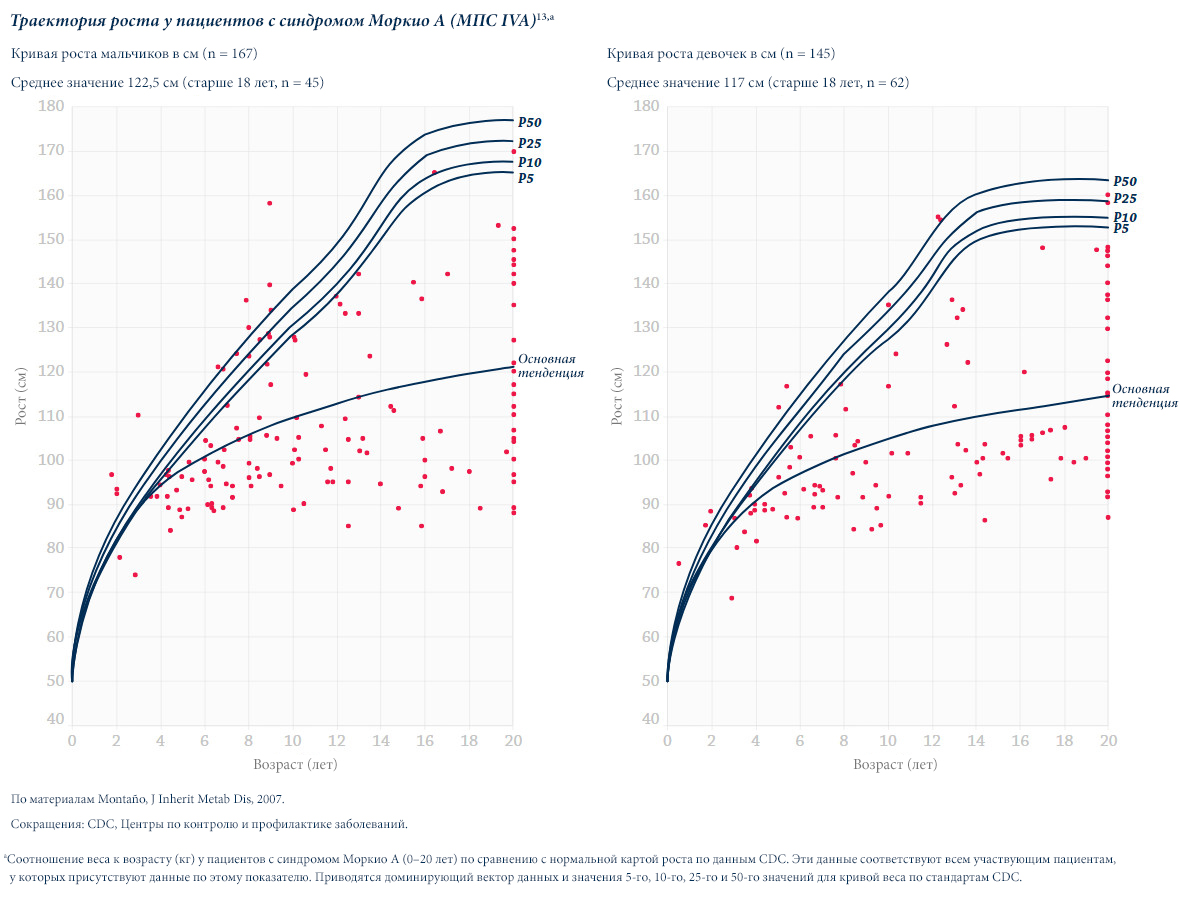
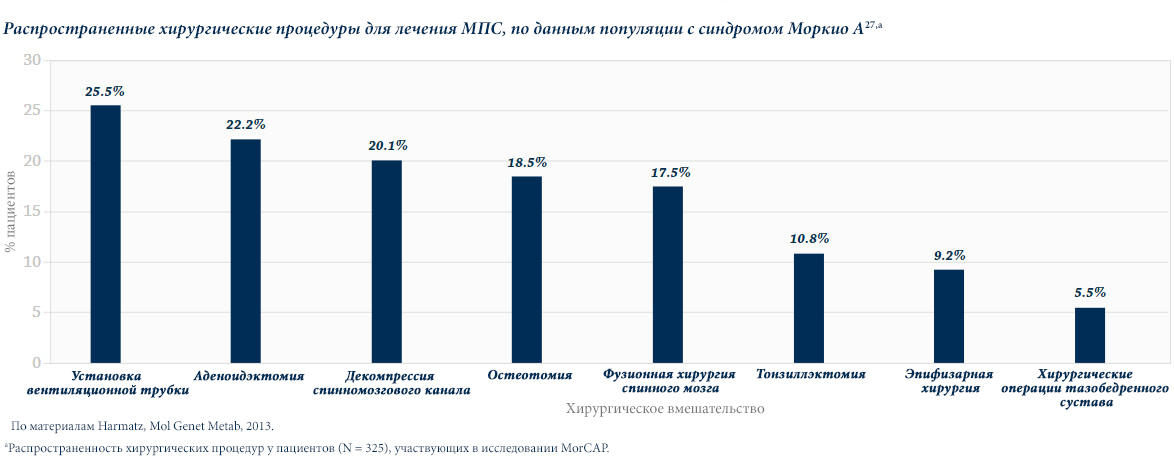
Различные виды МПС могут иметь сходные клинические проявления и скорость прогрессирования.Нижеприведенные рисунки иллюстрируют различные варианты прогрессирования МПС, наблюдаемые у пациентов с МПС VI типа.
У пациентов с МПС VI типа наблюдаются разные типы прогрессирования заболевания


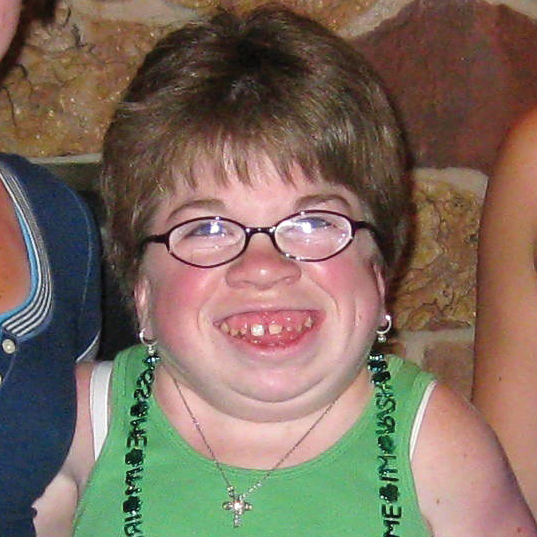
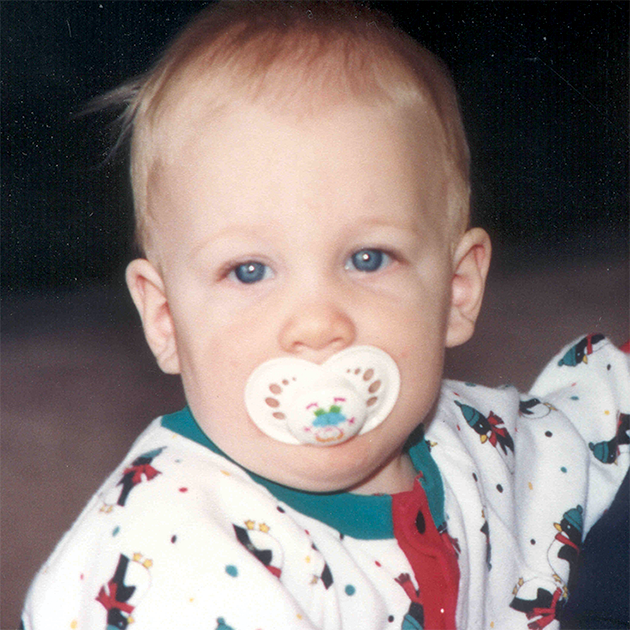

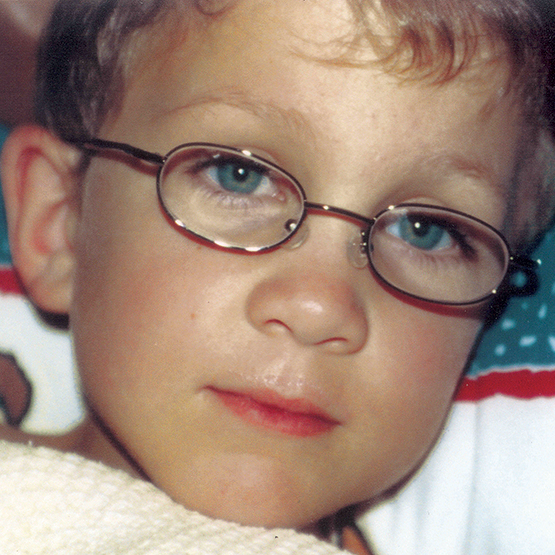



Задержка в постановке диагноза является частой проблемой и может иметь неблагоприятные последствия для пациентов3
Кажущаяся редкость заболевания, различия в характере прогрессирования и проявлениях заболевания, а также множество неспецифических симптомов, связанных с МПС, затрудняют диагностику. Период времени с момента появления первых симптомов до постановки диагноза может составлять от шести месяцев до десятков лет.3
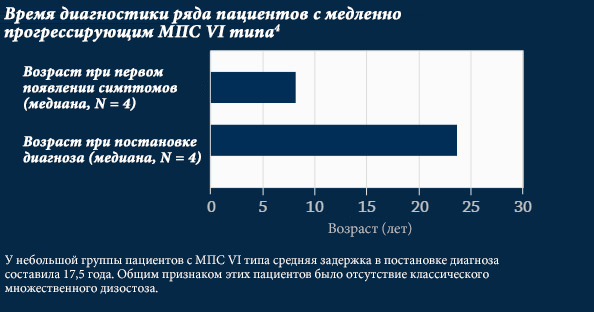
Ранняя диагностика имеет важное значение для улучшения прогноза лечения пациентов3,5
Ранняя диагностика может способствовать улучшению результатов лечения благодаря своевременному выбору соответствующей тактики ведения пациента, включающей ферментозаместительную терапию 3,5–8
Для многих мукополисахаридозов ФЗТ уже доступна или разрабатывается. Лучший способ диагностировать МПС и, тем самым, начать лечение — направить пациента с подозрением на заболевание к специалисту-генетику или в медико-генетический центр9
Обращайте внимание на ранние признаки и симптомы МПС, особенно:3,10–12
- Классические
- Грыжи
- Патологии суставов и скелета
- Низкий рост
- Любые постепенные, прогрессирующие изменения внешнего вида
- Неклассический фенотип
- Слабовыраженные проявления скелетных аномалий, включая утолщенные ребра или ключицы, слабовыраженная вальгусная деформация голени, патологии суставов или боль в суставах
- Пониженная переносимость физической нагрузки и/или необъяснимое сердечно-сосудистое поражение, в частности сердечный шум у пациентов детского возраста
- Необъяснимое снижение слуха или зрения (помутнение роговицы)